

Abstract
The receptor tyrosine kinase vascular endothelial growth factor (VEGF) receptor (VEGFR) plays an important role in tumor angiogenesis of hepatocellular carcinoma (HCC). (–)‐Epigallocatechin gallate (EGCG), the major biologically active component of green tea, inhibits growth in a variety of human cancer cells by inhibiting the activation of several types of receptor tyrosine kinases. In this study, we examined the effects of EGCG on the activity of the VEGF–VEGFR axis in human HCC cells. The levels of total and phosphorylated (i.e. activated) form of VEGFR‐2 protein (p‐VEGFR‐2) were observed to increase in a series of human HCC cell lines in comparison to the Hc normal human hepatocytes. EGCG preferentially inhibited the growth of HuH7 HCC cells, which express constitutive activation of the VEGF–VEGFR axis, in comparison to Hc cells. Treatment of HuH7 cells with EGCG caused a time‐ and dose‐dependent decrease in the expression of VEGFR‐2 and p‐VEGFR‐2 proteins. The production of VEGF from HuH7 cells was reduced by treatment with EGCG. Drinking of EGCG significantly inhibited the growth of HuH7 xenografts in nude mice and this was associated with inhibition of the activation of VEGFR‐2 and its related downstream signaling molecules, including ERK and Akt. EGCG drinking also decreased the expression of Bcl‐xL protein and VEGF mRNA in the xenografts. These findings suggest that EGCG can exert, at least in part, its growth‐inhibitive effect on HCC cells by inhibiting the VEGF–VEGFR axis. EGCG might therefore be useful in the treatment of HCC. (Cancer Sci 2009; 100: 1957–1962)
HCC, which commonly arises in the liver with chronic inflammation and cirrhosis, is a major health care problem worldwide.( 1 ) Because the prognosis of patients with HCC is poor, there is a critical need to develop more effective strategies for the therapy and prevention of this malignancy. Recent studies have revealed that the aberrant activation of several RTK and related downstream pathways of signal transduction play a critical role in the development of HCC and thus might be promising targets for the treatment of this cancer.( 2 , 3 , 4 ) For instance, sorafenib, a multikinase inhibitor that targets the serine‐threonine kinases Raf‐1 and B‐Raf and the RTK activity of VEGFR‐1, VEGFR‐2, and VEGFR‐3 and platelet‐derived growth factor receptor‐β, has shown a survival benefit in patients with HCC.( 5 ) In preclinical experiments, sorafenib exerted antiproliferative effects on the HCC‐derived cell lines and reduced tumor growth by inhibiting angiogenesis in a mouse xenograft model of human HCC.( 6 )
It is widely accepted that neovascularization and angiogenesis play a key role in the growth of solid tumor.( 7 , 8 ) VEGF, which binds to and activates VEGFR, is important in pathological angiogenesis, and the VEGF–VEGFR axis is therefore closely associated with tumor growth.( 7 , 8 ) In particular, HCC is a well‐known hypervascular tumor and a close relationship has been demonstrated between VEGF expression and either angiogenic activity or tumor progression in HCC.( 9 , 10 ) Overexpression of VEGFR is also observed in human HCC and this has been shown to correlate with a poor prognosis.( 11 , 12 ) In addition, several HCC cell lines express VEGFR and VEGF may act as an autocrine growth factor in stimulating the proliferation of these cells.( 13 ) These findings suggest that inhibition of the VEGF–VEGFR axis can theoretically reduce angiogenesis and tumor growth in HCC, and several agents that target this axis have been developed for the treatment of HCC.( 2 , 3 , 4 , 5 ) In a recent phase II study, bevacizumab, a humanized anti‐VEGF monoclonal antibody, alone or in combination with cytotoxic agents was used as treatment for patients with HCC and they showed a moderate antitumor activity.( 14 , 15 ) In HCC cells, RTK787, a tyrosine kinase inhibitor of VEGFR, inhibited tumor cell proliferation and induced apoptosis both in vivo and in vitro. ( 13 )
Numerous epidemiological and experimental studies suggest that green tea catechins have both anticancer and cancer chemopreventive effects at various organ sites.( 16 , 17 , 18 ) One of the anticancer mechanisms of green tea or its constituents is explained by their inhibitory effect on angiogenensis. Namely, EGCG, the major biologically active component of green tea, induces potent inhibition of VEGF‐dependent tyrosine phosphorylation of VEGFR‐2 and this is associated with the suppression of in vitro angiogenesis.( 19 ) The production of VEGF by cancer cells decreases after treatment with EGCG, thus contributing to its potent antiangiogenic activity.( 20 , 21 ) Green tea extract and EGCG also caused a decrease in VEGF production by HCC cells;( 22 ) however, whether EGCG can inhibit activation of the VEGF–VEGFR axis, thus inducing growth inhibition of HCC tumor, has not yet been examined. In the present study we investigated in detail the effects of EGCG on activation of the VEGF–VEGFR axis and the growth of HCC cells using in vitro and in vivo models.
Materials and Methods
Chemicals. EGCG was obtained from Mitusi Norin Co. (Tokyo, Japan).
Cell lines and cell culture conditions. Six human HCC cell lines, HLF, PLC/PRF/5, HepG2, HuH7, HLE, and Hep3B, were obtained from the Japanese Cancer Research Resources Bank (Tokyo, Japan) and maintained in DF10 medium containing DMEM (Invitrogen, San Diego, CA, USA) supplemented with 10% fetal bovine serum (Invitrogen). The Hc human normal hepatocyte cell line was purchased from Applied Cell Biology Research Institute (Kirkland, WA, USA) and maintained in CS‐C complete medium (Cell Systems Biotechnologie Vertrieb, St Katharinen, Germany). The cells were cultured in an incubator with humidified air with 5% CO2 at 37°C.
Cell viability assays. Cell viability assays were conducted using the MTT cell proliferation kit I (Roche Diagnosis Co., Indianapolis, IN, USA), according to the manufacturer's instructions, as described previously.( 23 ) Three thousand HuH7 or Hc cells were seeded into 96‐well plates. Twenty‐four hours later, the cells were treated with the indicated concentrations of EGCG (0–100 µg/mL) for 48 h in DF10 medium, and cell viability was examined. All assays were carried out in triplicate.
VEGF production assays. HuH7 cells were plated into six‐well plates and grown to 70% confluence. After washing with PBS, the cells were treated with the indicated concentrations of EGCG (0–100 µg/mL) in serum‐minus medium for 24 h. The cell‐free medium was then collected and the amounts of VEGF secreted by the cells into the medium were measured using a VEGF ELISA kit (R&D Systems, Minneapolis, MN, USA), according to the manufacturer's instructions.
Protein extraction and western blot analysis. Total protein was extracted from the cell lines or xenografts of HuH7 cells and equivalent amounts of protein (20 µg/lane) were subjected to a western blot analysis, as described previously.( 24 , 25 ) The primary antibodies for ERK, p‐ERK, Akt, and p‐Akt were described previously.( 23 ) The primary antibodies for VEGFR‐2 (#2479), p‐VEGFR‐2 (#2478), Bcl‐xL (#2762), and GAPDH (#2118) were purchased from Cell Signaling Technology (Beverly, MA, USA). An antibody to GAPDH served as a loading control.
RNA extraction and semiquantitative RT‐PCR analysis. A semiquantitative RT‐PCR analysis was carried out, as described previously.( 26 ) Total RNA was isolated from the xenopgrafts of HuH7 cells using ISOGEN reagent (Nippon Gene Co., Tokyo, Japan), according to the manufacturer's instructions. The cDNA was amplified from 1 µg of total RNA using SuperScript one‐step RT‐PCR with the platinum Taq system (Invitrogen). The primers used for amplification of VEGF and GAPDH specific genes were as follows: VEGF forward, 5′‐CTA CCT CCA CCA TGC CAA GT‐3′; VEGF reverse, 5′‐AAA TGC TTT CTC CGC TCT GA‐3′; GAPDH forward, 5′‐CGA GAT CCC TCC AAA ATC AA‐3′; and GAPDH reverse, 5′‐TTC AGC TCA GGG ATG ACC TT‐3′. Using a thermal controller (Programmable Thermal Controller; MJ Research, Watertown, MA, USA), 35‐cycle rounds of PCR were chosen for data analysis of the mRNA expression as a semiquantitative assessment indicated that the reaction had not reached a plateau and was still in log phase. The amplified products obtained with GAPDH‐specific primers served as an internal control. The intensities of the PCR products stained with ethidium bromide were quantified using the NIH Image software program version 1.62 (URL: http://rsb.info.nih.gov/nih‐image/index.html).
In vivo experimental protocol. Twenty‐four male BALB/c nude mice (5 weeks of age) were obtained from Charles River Japan (Tokyo, Japan). All mice were maintained at Gifu University Life Science Research Center, according to the Institutional Animal Care Guidelines, and were housed in plastic cages with free access to drinking water (tap water supplemented with or without EGCG) and the pelleted basal diet CRF‐1 (Oriental Yeast Co., Tokyo, Japan). Xenograft tumors were made by subcutaneous injection of HuH7 cells, at a concentration of 5 × 106 cells per 200 µL, into the flanks of these mice. One week after tumor cell injection, the mice were randomly divided into three groups (eight mice per group) and then treated with (groups 2 and 3) or without (group 1) EGCG for 5 weeks. The mice in groups 2 and 3 were given tap water containing 0.01 or 0.1% EGCG, respectively. The concentrations of EGCG (0.01 and 0.1%) were established according to the findings of previous reports( 27 , 28 ) because these doses could exert anticancer properties without causing various side effects in any organs. The mice in group 1 were given tap water and served as an untreated control. A freshly prepared solution of EGCG in tap water was supplied to the experimental mice three times a week. The tumor size and bodyweight were measured once a week and the tumor volume was calculated using the formula: largest diameter × (smallest diameter)2 × 0.5.
Statistical analysis. The data were expressed as the mean ± SD. The statistical significance of the difference in mean values was tested using a one‐way analysis of variance (ANOVA) and the unpaired t‐test. Significance was defined as a P‐value less than 0.05. All analyses were carried out using the StatView ver. 5.0 software program (SAS Institute, Cary, NC, USA).
Results
Expression of VEGFR‐2 and p‐VEGFR‐2 proteins in human HCC cell lines and Hc normal hepatocytes. Among the VEGFR, VEGFR‐2 is considered to be the major mediator of the mitogenic and angiogenic effects of VEGF.( 7 , 8 ) We therefore initially examined whether VEGFR‐2 protein is overexpressed and constitutively activated in HLF, PLC/PRF/5, HepG2, HuH7, HLE, and Hep3B human HCC cell lines and in Hc human normal hepatocytes using western blot analysis. Among these HCC cell lines, the level of VEGFR‐2 protein was observed to markedly increase in the HLF and HLE cells, whereas it was moderately expressed in Hep3B and HuH7 cells (Fig. 1). The level of phosphorylated (i.e. activated) VEGFR‐2 protein (p‐VEGFR‐2) also increased in these four cell lines, thus indicating the constitutive activation of this receptor (Fig. 1). Moreover, all HCC cell lines that were examined in this experiment significantly expressed VEGFR‐2 and p‐VEGFR‐2 proteins in comparison to the Hc normal human hepatocytes (Fig. 1).
Figure 1.
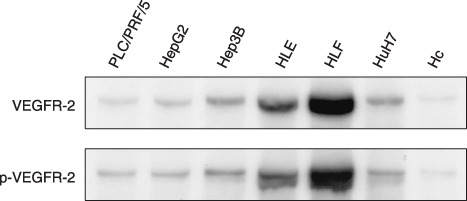
The expression levels of total vascular endothelial growth factor receptor (VEGFR)‐2 and phosphorylated vascular endothelial growth factor receptor (p‐VEGFR)‐2 proteins in human hepatocellular carcinoma cell lines and Hc normal hepatocytes. Total protein extracts were prepared from 70% confluent cultures of the indicated cell lines and equivalent amounts of protein (20 µg/lane) were examined by western blot analysis using appropriate antibodies. Repeat western blots gave similar results.
Effects of EGCG on the growth of HuH7 and Hc cells. We then examined the growth‐inhibitory effects of EGCG on HuH7 and Hc cell lines using MTT assays. As shown in Figure 2, EGCG inhibited the growth of HuH7 cells with an IC50 value of approximately 25 µg/mL. However, the Hc cells were more resistant to EGCG because the IC50 value with this agent was approximately 84 µg/mL (Fig. 2). These findings suggest that EGCG preferentially inhibits the growth of HuH7 HCC cells, which express higher levels of the VEGFR‐2 and p‐VEGFR‐2 proteins, when compared with Hc cells that do not express these proteins (Fig. 1).
Figure 2.
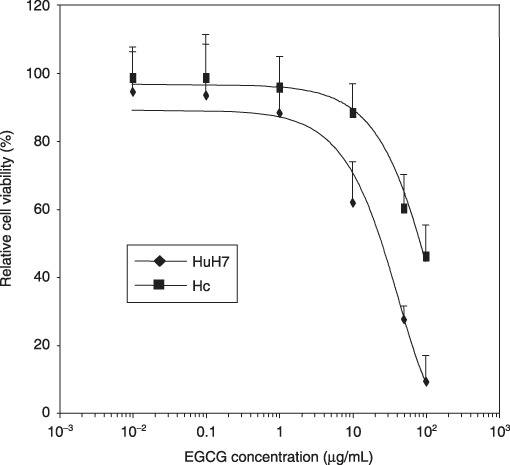
Inhibition of cell growth by (–)‐epigallocatechin gallate (EGCG) in HuH7 human hepatocellular carcinoma cells and Hc normal hepatocytes. These cells were treated with the indicated concentrations of EGCG or DMSO for 48 h and cell viability assays were conducted using the MTT system. Results are expressed as a percentage of growth with 100% representing control cells treated with DMSO alone. Bars, SD of triplicate assays.
Effects of EGCG on the expression and activation of VEGFR‐2 in HuH7 cells. We next examined whether EGCG alters the expression and activation of VEGFR‐2 in HuH7 cells. A time‐course study indicated that when the cells were treated with 25 µg/mL EGCG, which is the same as the IC50 concentration determined by the MTT assays (Fig. 2), a marked decrease was observed in the expression levels of both VEGFR‐2 and p‐VEGFR‐2 proteins within 6 h of the addition of this agent (Fig. 3a). When the cells were treated with the indicated concentrations of EGCG (0–100 µg/mL) for 6 h, the expression levels of VEGFR‐2 as well as p‐VEGFR‐2 proteins were also inhibited in a dose‐dependent manner (Fig. 3b).
Figure 3.
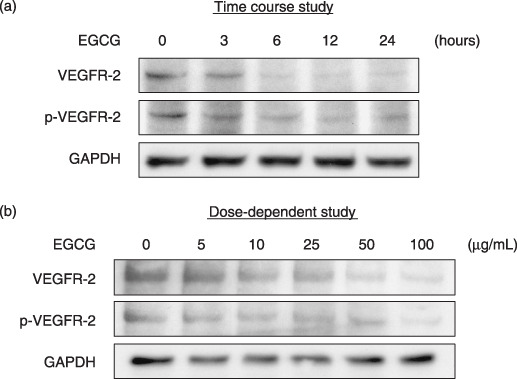
Effects of (–)‐epigallocatechin gallate (EGCG) on expression levels of total vascular endothelial growth factor receptor (VEGFR)‐2 and phosphorylated vascular endothelial growth factor receptor (p‐VEGFR)‐2 proteins in HuH7. The cells were treated with (a) 25 µg/mL EGCG for the indicated times (0, 3, 6, 12, and 24 h, time course study) or (b) the indicated concentration of EGCG (0, 5, 10, 25, 50, and 100 µg/mL, dose‐dependence study) for 6 h, and the cell extracts were then examined by western blot analysis using the respective antibodies. An antibody to GAPDH served as a loading control. Similar results were obtained in a repeat experiment.
Effects of EGCG on VEGF production by HuH7 cells. VEGF, which is produced by cancer cells, has been reported to play a critical role in tumor angiogenesis.( 7 , 8 ) We therefore next examined the effects of EGCG on production of VEGF by HuH7 cells using an ELISA system. As shown in Figure 4, HuH7 cells secreted an abundant amount of VEGF into the growth medium when the cells were cultured in serum‐free medium for 24 h and, interestingly, a low (10 µg/mL, less than IC50 value) and high (100 µg/mL) concentration of EGCG significantly reduced the production of VEGF from these cancer cells.
Figure 4.
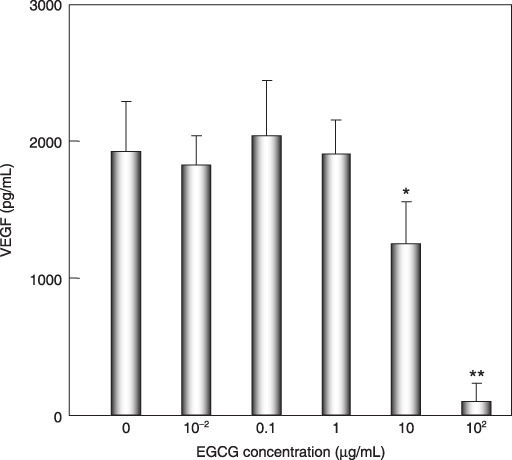
Effects of (–)‐epigallocatechin gallate (EGCG) on production of vascular endothelial growth factor (VEGF) by HuH7 cells. The cells were treated with the indicated concentration of EGCG (0, 0.01, 0.1, 1.0, 10, and 100 µg/mL) in serum‐free medium for 24 h. The medium was then collected and assayed for VEGF using an ELISA kit. Bars, SD of triplicate assays. *P < 0.05, **P < 0.01: significant differences obtained by comparison with EGCG‐untreated control group.
Effects of EGCG on the growth of HCC xenografts in nude mice. We next examined whether the growth inhibition of the treatment with EGCG in HuH7 cells was also observed in vivo using a nude mouse xenograft model. Figure 5 shows that drinking water with not only a high concentration (0.1%), but also a low concentration (0.01%) of EGCG strongly inhibited the growth of the HuH7 xenograft during treatment with this agent (5 weeks). The tumor volume of the mice treated with 0.1% EGCG was less than that of tumors in 0.01% EGCG‐drinking mice, but the difference was not significant (Fig. 5). All of the treatments were well tolerated and the bodyweights remained stable in all groups during the experiment (data not shown). There were no pathological alterations suggesting toxicity of EGCG in the liver, spleen, and kidneys of mice (data not shown).
Figure 5.
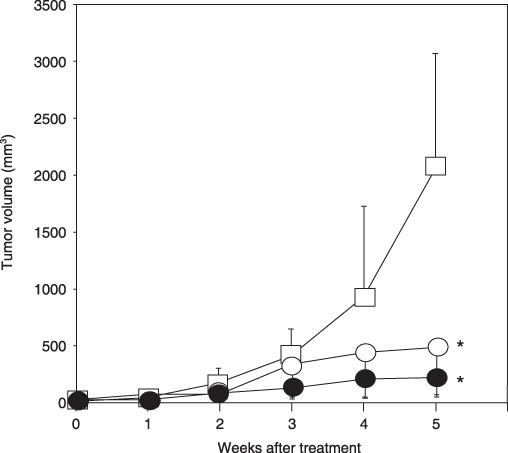
Effects of (–)‐epigallocatechin gallate (EGCG) on the growth of HuH7 xenografts in nude mice. Male BALB/c nude mice were injected subcutaneously with 5 × 106 HuH7 cells. One week after the injection, the mice were divided into three groups and treated with following conditions for 5 weeks: group 1, control group (tap water drinking group,  ); group 2, 0.01% EGCG‐drinking group (
); group 2, 0.01% EGCG‐drinking group ( ); and group 3, 0.1% EGCG‐drinking group (
); and group 3, 0.1% EGCG‐drinking group ( ). The growth curve of HuH7 tumors in each group are represented. Bars, SD. *P < 0.05: significant differences obtained by comparison with EGCG‐untreated control group.
). The growth curve of HuH7 tumors in each group are represented. Bars, SD. *P < 0.05: significant differences obtained by comparison with EGCG‐untreated control group.
Effects of EGCG on the activation of VEGFR‐2 and its downstream signaling molecules and on the cellular levels of Bcl‐xL in xenografts of HuH7 cells. We next examined whether treatment with EGCG inhibits the activation of VEGFR‐2 and its multiple downstream signaling pathways in the HuH7 xenografts. Drinking both 0.01 and 0.1% EGCG decreased the total levels of VEGFR‐2 and Akt proteins in these xenografts (Fig. 6a). There was also a marked decrease in the levels of p‐VEGFR‐2, p‐ERK, and p‐Akt proteins by treatment with both concentrations of EGCG (Fig. 6a). In addition, EGCG caused a decrease in the levels of Bcl‐xL, an anti‐apoptotic Bcl‐2 family member, in HuH7 xenogrfts (Fig. 6a).
Figure 6.
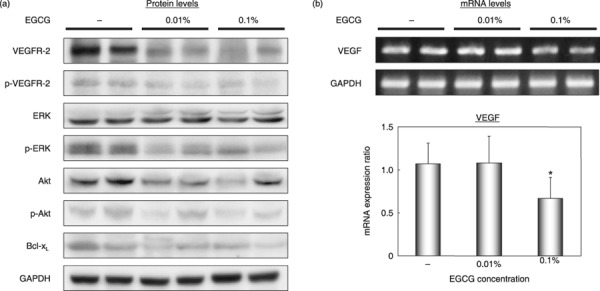
Effects of (–)‐epigallocatechin gallate (EGCG) on activation of vascular endothelial growth factor receptor (VEGFR)‐2, its related downstream signaling pathways, and on the cellular levels of Bcl‐xL proteins and vascular endothelial growth factor (VEGF) mRNA in HuH7 xenografts. The xenografts were excised from each animal at the termination of the experiment and tumor extracts were examined by (a) western blot analysis using the respective antibodies or (b) a semiquantitative RT‐PCR analysis using VEGF‐specific primers. An antibody to GAPDH served as a loading control (A). Amplified PCR products obtained with GAPDH‐specific primers served as internal controls. (b) The results obtained from RT‐PCR analysis were quantified by densitometry and are displayed in the lower panel. Bars, SD of triplicate assays. *P < 0.05: significant differences obtained by comparison with EGCG‐untreated control group. p‐, phosphorylated.
Effects of EGCG on the expression of VEGF mRNA in xenografts of HuH7 cells. A semiquantitative RT‐PCR study showed that there was a significant decrease in the level of VEGF mRNA in the xenografts of mice treated with 0.1% EGCG compared to that of the tumors in control mice (Fig. 6b). Therefore, the inhibitory effect of EGCG on the production of VEGF was not only observed in vitro (Fig. 4) but also in vivo.
Discussion
The results of the present study clearly indicate that EGCG effectively suppresses the growth of HuH7 human HCC cells both in vitro (Fig. 2) and in vivo (Fig. 5) and this was associated with inhibition of the VEGF–VEGFR axis (3, 6). It should be particularly emphasized that not only a high concentration (0.1%), but also a low concentration (0.01%) of EGCG similarly inhibited the growth of HCC xenografts by blocking the VEGF–VEGFR axis to the same extent (5, 6). This is the first report indicating a low dose of EGCG (0.01%) to be sufficient to reduce HCC tumor growth, although the feeding protocol of EGCG at a high dose (0.1%), which mimics an approximate consumption of six cups of green tea per day by an average adult human, has been used in mice in many prior chemopreventive studies.( 16 , 29 ) These findings, together with the result of a previous study reporting that drinking 0.05% EGCG also significantly repressed the tumor growth of highly angiogenic sarcoma xenografts by inhibiting angiogenesis in vivo,( 30 ) might thus be preferable when considering the clinical use of this agent because a lower dose is more acceptable for administration to patients.
One of the main mechanisms of how EGCG can block the VEGF–VEGFR axis in cancer cells is explained by its efficacy in reducing VEGF secretion from these cells.( 20 , 21 , 22 ) The expression of VEGF is regulated by micro‐environmental alterations, such as hypoxia, and recent studies have revealed that HIF‐1 strongly activates transcription of the VEGF gene by phosphorylating the ERK and Akt proteins.( 7 , 31 , 32 , 33 ) The increased expression of VEGFR and HIF‐1α, the regulated subunit of HIF‐1, is considered to play a role in the progression of HCC.( 34 ) Hepatitis C virus infection leads to the stabilization of HIF‐1α via activation of the MAPK–ERK and PI3K–Akt signaling pathways, thus leading to neovascularization.( 35 ) In the present study we demonstrated that EGCG reduces the expression of VEGF mRNA and production of this growth factor by inhibiting the activation of ERK and Akt proteins in HuH7 cells (4, 6). These findings are consistent with those of a previous report, in which green tea extract and EGCG were observed to cause a drastic decrease in VEGF expression at both the mRNA and protein levels by suppressing the expression of HIF‐1α and blocking both the PI3K–Akt and MAPK–ERK signaling pathways in HepG2 human HCC cells.( 22 ) Because several HCC cell lines express the constitutive activation of VEGFR‐2 (Fig. 1), our findings, together with those of the previous report,( 22 ) suggest the possibility that EGCG might be able to inhibit cell growth by disrupting the VEGF–VEGFR‐related autocrine loop that exists in HCC cells.
The transcription of VEGF mRNA is also induced by the activation of a variety of RTK.( 7 ) For instance, activation of the IGF‐1–IGF‐1 receptor axis induces expression of the VEGF gene via induction of HIF‐1α.( 31 ) Both the activation of EGFR and HER2 induce the secretion of VEGF by activating the PI3K–Akt signaling pathway.( 32 , 33 ) These reports seem to be significant when considering the characteristic effects of EGCG because this agent can inhibit not only VEGFR‐2 (3, 6), but also the activation of several other RTK, including IGF‐1R, EGFR, HER2, and HER3, and multiple downstream signaling pathways in human HCC and colon cancer cells.( 23 , 24 , 25 , 26 ) One of the possible mechanisms for this remarkable range of effects by EGCG might be associated with its ability to bind directly to all of these receptors, thereby inhibiting their tyrosine kinase activities, perhaps because of sufficient homologies in their kinase domain.( 36 ) This presumption might be supported by the previous report that EGCG can act directly to inhibit the kinase activity of some RTK.( 37 , 38 ) The recent report describing that EGCG inhibits the binding of VEGF to VEGFR in a concentration‐dependent manner( 39 ) also encourages this hypothesis. In addition, the effects of EGCG to decrease the total levels of VEGFR‐2 itself contribute to inhibit activation of the VEGF–VEGFR axis in the present study (3, 6). These findings are consistent with a recent report that green tea extract could decrease the expression of both VEGFR‐1 and VEGFR‐2 on human umbilical vein endothelial cells.( 40 )
In addition to the direct effects of EGCG on specific RTK at the cell surface, recent studies revealed that EGCG exerts its effects on RTK indirectly by targeting the lipid organization of the plasma membrane, so‐called ‘lipid rafts’, which are associated with these RTK. Indeed, the inhibitory effect of EGCG on EGF binding to the EGFR and the subsequent dimerization of this receptor is associated with alterations in the lipid rafts of colon cancer cells.( 41 ) EGCG also decreases cell surface‐associated EGFR by inducing the internalization of EGFR into endosomal vesicles, thereby inhibiting the activation of this receptor and exerting anticancer effects.( 42 ) Because VEGFR‐2 is also localized to lipid rafts,( 43 ) future studies would be required to elucidate whether the inhibitory effect of EGCG on the activation of VEGFR‐2 is associated with alterations in membrane lipid order in cancer cells.
Agents that can co‐target several different RTK, such as sorafenib, are expected to be promising candidates for the treatment of HCC.( 2 , 3 , 4 , 5 , 6 ) In conclusion, the ability of EGCG to target both the VEGF–VEGFR axis, as demonstrated in the present study, and other types of RTK that play critical roles in the proliferation of cancer cells,( 23 , 24 , 25 , 26 ) is thus considered to provide evidence that this naturally occurring agent may be effective in the chemoprevention and therapy of HCC, and likely of other malignancies as well, that show hypervascularity.
Abbreviations
| EGCG | (–)‐epigallocatechin gallate |
| EGFR | epidermal growth factor receptor |
| ERK | extracellular signal‐regulated kinase |
| HCC | hepatocellular carcinoma |
| HER | human epidermal growth factor receptor |
| HIF | hypoxia‐inducible factor |
| IGF | insulin like growth factor |
| PDGFR | platelet‐derived growth factor receptor |
| RTK | receptor tyrosine kinase |
| VEGF | vascular endothelial growth factor |
| VEGFR | vascular endothelial growth factor receptor. |
Acknowledgments
This work was supported in part by Grants‐in‐Aid from the Ministry of Education, Science, Sports, and Culture of Japan (no. 18790457 to M.S. and no. 17015016 to H.M.).
References
- 1. El‐Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology 2007; 132: 2557–76. [DOI] [PubMed] [Google Scholar]
- 2. Llovet JM, Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology 2008; 48: 1312–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pang RW, Poon RT. From molecular biology to targeted therapies for hepatocellular carcinoma: the future is now. Oncology 2007; 72(Suppl 1): 30–44. [DOI] [PubMed] [Google Scholar]
- 4. Tanaka S, Arii S. Molecularly targeted therapy for hepatocellular carcinoma. Cancer Sci 2009; 100: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Llovet JM, Ricci S, Mazzaferro V et al . Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008; 359: 378–90. [DOI] [PubMed] [Google Scholar]
- 6. Liu L, Cao Y, Chen C et al . Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res 2006; 66: 11 851–8. [DOI] [PubMed] [Google Scholar]
- 7. Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med 2003; 9: 669–76. [DOI] [PubMed] [Google Scholar]
- 8. Ellis LM, Hicklin DJ. VEGF‐targeted therapy: mechanisms of anti‐tumour activity. Nature Rev Cancer 2008; 8: 579–91. [DOI] [PubMed] [Google Scholar]
- 9. Mise M, Arii S, Higashituji H et al . Clinical significance of vascular endothelial growth factor and basic fibroblast growth factor gene expression in liver tumor. Hepatology 1996; 23: 455–64. [DOI] [PubMed] [Google Scholar]
- 10. Torimura T, Sata M, Ueno T et al . Increased expression of vascular endothelial growth factor is associated with tumor progression in hepatocellular carcinoma. Hum Pathol 1998; 29: 986–91. [DOI] [PubMed] [Google Scholar]
- 11. Ng IO, Poon RT, Lee JM, Fan ST, Ng M, Tso WK. Microvessel density, vascular endothelial growth factor and its receptors Flt‐1 and Flk‐1/KDR in hepatocellular carcinoma. Am J Clin Pathol 2001; 116: 838–45. [DOI] [PubMed] [Google Scholar]
- 12. Dhar DK, Naora H, Yamanoi A et al . Requisite role of VEGF receptors in angiogenesis of hepatocellular carcinoma: a comparison with angiopoietin/Tie pathway. Anticancer Res 2002; 22: 379–86. [PubMed] [Google Scholar]
- 13. Liu Y, Poon RT, Li Q, Kok TW, Lau C, Fan ST. Both antiangiogenesis‐ and angiogenesis‐independent effects are responsible for hepatocellular carcinoma growth arrest by tyrosine kinase inhibitor PTK787/ZK222584. Cancer Res 2005; 65: 3691–9. [DOI] [PubMed] [Google Scholar]
- 14. Siegel AB, Cohen EI, Ocean A et al . Phase II trial evaluating the clinical and biologic effects of bevacizumab in unresectable hepatocellular carcinoma. J Clin Oncol 2008; 26: 2992–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhu AX, Blaszkowsky LS, Ryan DP et al . Phase II study of gemcitabine and oxaliplatin in combination with bevacizumab in patients with advanced hepatocellular carcinoma. J Clin Oncol 2006; 24: 1898–903. [DOI] [PubMed] [Google Scholar]
- 16. Yang CS, Maliakal P, Meng X. Inhibition of carcinogenesis by tea. Annu Rev Pharmacol Toxicol 2002; 42: 25–54. [DOI] [PubMed] [Google Scholar]
- 17. Shimizu M, Weinstein IB. Modulation of signal transduction by tea catechins and related phytochemicals. Mutat Res 2005; 591: 147–60. [DOI] [PubMed] [Google Scholar]
- 18. Khan N, Afaq F, Saleem M, Ahmad N, Mukhtar H. Targeting multiple signaling pathways by green tea polyphenol (–)‐epigallocatechin‐3‐gallate. Cancer Res 2006; 66: 2500–5. [DOI] [PubMed] [Google Scholar]
- 19. Lamy S, Gingras D, Beliveau R. Green tea catechins inhibit vascular endothelial growth factor receptor phosphorylation. Cancer Res 2002; 62: 381–5. [PubMed] [Google Scholar]
- 20. Masuda M, Suzui M, Lim JT, Deguchi A, Soh JW, Weinstein IB. Epigallocatechin‐3‐gallate decreases VEGF production in head and neck and breast carcinoma cells by inhibiting EGFR‐related pathways of signal transduction. J Exp Ther Oncol 2002; 2: 350–9. [DOI] [PubMed] [Google Scholar]
- 21. Jung YD, Kim MS, Shin BA et al . EGCG, a major component of green tea, inhibits tumour growth by inhibiting VEGF induction in human colon carcinoma cells. Br J Cancer 2001; 84: 844–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang Q, Tang X, Lu Q, Zhang Z, Rao J, Le AD. Green tea extract and (–)‐epigallocatechin‐3‐gallate inhibit hypoxia‐ and serum‐induced HIF‐1alpha protein accumulation and VEGF expression in human cervical carcinoma and hepatoma cells. Mol Cancer Ther 2006; 5: 1227–38. [DOI] [PubMed] [Google Scholar]
- 23. Shimizu M, Deguchi A, Lim JT, Moriwaki H, Kopelovich L, Weinstein IB. (–)‐Epigallocatechin gallate and polyphenon E inhibit growth and activation of the epidermal growth factor receptor and human epidermal growth factor receptor‐2 signaling pathways in human colon cancer cells. Clin Cancer Res 2005; 11: 2735–46. [DOI] [PubMed] [Google Scholar]
- 24. Shimizu M, Deguchi A, Joe AK, McKoy JF, Moriwaki H, Weinstein IB. EGCG inhibits activation of HER3 and expression of cyclooxygenase‐2 in human colon cancer cells. J Exp Ther Oncol 2005; 5: 69–78. [PubMed] [Google Scholar]
- 25. Shimizu M, Deguchi A, Hara Y, Moriwaki H, Weinstein IB. EGCG inhibits activation of the insulin‐like growth factor‐1 receptor in human colon cancer cells. Biochem Biophys Res Commun 2005; 334: 947–53. [DOI] [PubMed] [Google Scholar]
- 26. Shimizu M, Shirakami Y, Sakai H et al . EGCG inhibits activation of the insulin‐like growth factor (IGF)/IGF‐1 receptor axis in human hepatocellular carcinoma cells. Cancer Lett 2008; 262: 10–18. [DOI] [PubMed] [Google Scholar]
- 27. Shimizu M, Shirakami Y, Sakai H et al . (–)‐Epigallocatechin gallate suppresses azoxymethane‐induced colonic premalignant lesions in male C57BL/KsJ‐db/db mice. Cancer Prev Res 2008; 1: 298–304. [DOI] [PubMed] [Google Scholar]
- 28. Shirakami Y, Shimizu M, Tsurumi H, Hara Y, Tanaka T, Moriwaki H. EGCG and Polyphenon E attenuate inflammation‐related mouse colon carcinogenesis induced by AOM plus DDS. Mol Med Rep 2008; 1: 355–61. [PubMed] [Google Scholar]
- 29. Wang ZY, Agarwal R, Bickers DR, Mukhtar H. Protection against ultraviolet B radiation‐induced photocarcinogenesis in hairless mice by green tea polyphenols. Carcinogenesis 1991; 12: 1527–30. [DOI] [PubMed] [Google Scholar]
- 30. Fassina G, Vene R, Morini M et al . Mechanisms of inhibition of tumor angiogenesis and vascular tumor growth by epigallocatechin‐3‐gallate. Clin Cancer Res 2004; 10: 4865–73. [DOI] [PubMed] [Google Scholar]
- 31. Fukuda R, Hirota K, Fan F, Jung YD, Ellis LM, Semenza GL. Insulin‐like growth factor 1 induces hypoxia‐inducible factor 1‐mediated vascular endothelial growth factor expression, which is dependent on MAP kinase and phosphatidylinositol 3‐kinase signaling in colon cancer cells. J Biol Chem 2002; 277: 38 205–11. [DOI] [PubMed] [Google Scholar]
- 32. Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signaling increases the rate of hypoxia‐inducible factor 1alpha (HIF‐1alpha) synthesis: novel mechanism for HIF‐1‐mediated vascular endothelial growth factor expression. Mol Cell Biol 2001; 21: 3995–4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhong H, Chiles K, Feldser D et al . Modulation of hypoxia‐inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3‐kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res 2000; 60: 1541–5. [PubMed] [Google Scholar]
- 34. Nakamura K, Zen Y, Sato Y et al . Vascular endothelial growth factor, its receptor Flk‐1, and hypoxia inducible factor‐1alpha are involved in malignant transformation in dysplastic nodules of the liver. Hum Pathol 2007; 38: 1532–46. [DOI] [PubMed] [Google Scholar]
- 35. Nasimuzzaman M, Waris G, Mikolon D, Stupack DG, Siddiqui A. Hepatitis C virus stabilizes hypoxia‐inducible factor 1alpha and stimulates the synthesis of vascular endothelial growth factor. J Virol 2007; 81: 10 249–57. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36. Blume‐Jensen P, Hunter T. Oncogenic kinase signalling. Nature 2001; 411: 355–65. [DOI] [PubMed] [Google Scholar]
- 37. Liang YC, Lin‐shiau SY, Chen CF, Lin JK. Suppression of extracellular signals and cell proliferation through EGF receptor binding by (–)‐epigallocatechin gallate in human A431 epidermoid carcinoma cells. J Cell Biochem 1997; 67: 55–65. [DOI] [PubMed] [Google Scholar]
- 38. Sachinidis A, Seul C, Seewald S, Ahn H, Ko Y, Vetter H. Green tea compounds inhibit tyrosine phosphorylation of PDGF beta‐receptor and transformation of A172 human glioblastoma. FEBS Lett 2000; 471: 51–5. [DOI] [PubMed] [Google Scholar]
- 39. Kondo T, Ohta T, Igura K, Hara Y, Kaji K. Tea catechins inhibit angiogenesis in vitro, measured by human endothelial cell growth, migration and tube formation, through inhibition of VEGF receptor binding. Cancer Lett 2002; 180: 139–44. [DOI] [PubMed] [Google Scholar]
- 40. Kojima‐Yuasa A, Hua JJ, Kennedy DO, Matsui‐Yuasa I. Green tea extract inhibits angiogenesis of human umbilical vein endothelial cells through reduction of expression of VEGF receptors. Life Sci 2003; 73: 1299–313. [DOI] [PubMed] [Google Scholar]
- 41. Adachi S, Nagao T, Ingolfsson HI et al . The inhibitory effect of (–)‐epigallocatechin gallate on activation of the epidermal growth factor receptor is associated with altered lipid order in HT29 colon cancer cells. Cancer Res 2007; 67: 6493–501. [DOI] [PubMed] [Google Scholar]
- 42. Adachi S, Nagao T, To S et al . (–)‐Epigallocatechin gallate causes internalization of the epidermal growth factor receptor in human colon cancer cells. Carcinogenesis 2008; 29: 1986–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Labrecque L, Royal I, Surprenant DS, Patterson C, Gingras D, Beliveau R. Regulation of vascular endothelial growth factor receptor‐2 activity by caveolin‐1 and plasma membrane cholesterol. Mol Biol Cell 2003; 14: 334–47. [DOI] [PMC free article] [PubMed] [Google Scholar]