

Abstract
Glycogen synthase kinase-3β (GSK-3β) is a central component in many critical intracellular signaling mechanisms. These include the phosphatidylinositol 3-kinase/Akt cell survival pathway, which inhibits GSK-3β activity. GSK-3β itself inhibits the activation of several transcription factors, which are important cell survival factors, such as heat shock factor 1. These factors likely contribute to the recent revelation that GSK-3β is a pro-apoptotic enzyme. Recently, lithium has been identified as a selective and direct inhibitor of GSK-3β. Based on these findings, we have proposed that part of the neuroprotectant properties of lithium is due to its ability to inhibit GSK-3β, and thus block the facilitation of apoptosis produced by GSK-3β. Since several anticonvulsants recently have been shown to be effective mood stabilizers, we examined if these agents are capable of protecting cells from GSK-3β-facilitated apoptosis. In addition to lithium, both valproic acid and lamotrigine, but not carbamazepine, provided protection from GSK-3β-facilitated apoptosis in human neuroblastoma SH-SY5Y cells. These results demonstrate that several drugs therapeutic for bipolar disorder can provide neuroprotection by inhibiting the pro-apoptotic effects of GSK-3β, providing new evidence that dysregulation of GSK-3β may contribute to the pathophysiology of bipolar disorder.
Keywords: apoptosis, glycogen synthase kinase-3β, lamotrigine, lithium, neuroprotection, valproate
Evidence obtained from a diverse array of investigations completed during the last few years indicates that the therapeutic actions of lithium may arise from its effects on multiple targets (1). One component of this is evidence that lithium is a neuroprotectant, supporting neuronal survival in the face of potentially lethal insults. At least a part of the neuroprotection provided by lithium stems from its ability to inhibit the activity of glycogen synthase kinase-3β (GSK-3β), an enzyme recently found to contribute to apoptosis. This article briefly reviews these background topics, and then presents new results indicating that other mood stabilizers, in addition to lithium, also protect neurons from GSK-3β-facilitated apoptosis.
GSK-3β: regulation and cell fate
There is mounting evidence that alterations of GSK-3β activity can influence cell fate, emphasizing the need for strict cellular regulation of its activity. The activity of GSK-3β is controlled by signaling systems that are also associated with the regulation of cell growth and survival. For example, although GSK-3β is constitutively active, it can be activated by transient increases in calcium (2). GSK-3β can also be inhibited by signaling systems, such as the Wnt pathway (3), and by phosphorylation via the phosphatidylinositol 3-kinase/Akt (PI-3K/Akt) signaling pathway (4). These and other intracellular signaling pathways serve to maintain GSK-3 in an optimal state, and recent reports indicate that these mechanisms regulating GSK-3β may be important in controlling cell survival.
A direct link between GSK-3β activity and apoptosis has been revealed in several studies. The first evidence that activation of GSK-3β could be associated with neuronal apoptosis was the finding that the administration of antisense oligonucleotides to reduce GSK-3β levels protected cells from amyloid β-peptide-induced neurotoxicity (5). More recently, transient overexpression of GSK-3β was found to induce spontaneous apoptosis in rat PC12 cells in a caspase-3-dependent manner (6). Furthermore, overexpression of a catalytically inactive form of GSK-3β reduced apoptosis induced by treatment with an inhibitor of the PI-3K/Akt signaling pathway (6). These findings not only revealed that GSK-3β itself is capable of facilitating apoptosis, but also indicated that one mechanism by which the PI-3K/Akt signaling pathway promotes cell survival may be by down-regulating the activity of GSK-3β (6). In subsequent studies, Bijur et al. (7) reported that stable, moderately overexpressed GSK-3β in human neuroblastoma cells did not cause spontaneous apoptosis, but greatly facilitated apoptosis induced by treatment with staurosporine or heat shock, emphasizing the critical importance of properly controlling the activity of GSK-3β. Additionally, Hetman et al. (8) demonstrated the involvement of GSK-3β in apoptosis induced by serum withdrawal and treatment with an NMDA receptor antagonist or inhibitors of PI-3K in rat cortical neurons. Altogether, these findings indicate that GSK-3β has a regulatory role in neuronal apoptosis, indicating that control of GSK-3β activity is important for cell survival. Further studies aimed at clarifying the effects of cell stress on the activity of GSK-3β should be useful for advancing the understanding of how GSK-3β is regulated under conditions that can potentially cause cell death.
As noted above, GSK-3β activity is influenced by the activity of the PI-3K/Akt signaling pathway. Activation of the PI-3K/Akt signaling pathway commences when insulin, or growth factors, such as insulin-like growth factor, epidermal growth factor, and nerve growth factor, bind their respective receptors. Receptor stimulation results in the activation of PI-3K, which catalyzes the production of phosphatidylinositol 3,4-bisphosphate and phosphatidylinositol 3,4,5-trisphosphate, which subsequently is bound by the pleckstrin homology domain of Akt to bring it into close proximity with phosphoinositide-dependent kinase-1. This facilitates the phosphorylation of Akt on Thr308 by phosphoinositide-dependent kinase-1, and on Ser473 by a currently unknown kinase (9). Subsequently, Akt can phosphorylate Ser9 of GSK-3β, which inhibits its activity. Thus, activation of the PI-3K/Akt signaling pathway and the subsequent inhibition of GSK-3β is one mechanism by which growth factors may confer neuroprotection against pro-apoptotic stimuli.
Besides growth factors, Akt can be activated by toxic conditions, such as oxidative stress or by exposing cells to elevated temperatures, commonly known as ‘heat shock’. Stress-induced activation of Akt was initially described by Konishi et al. (10). Their study suggested that stress-induced activation of Akt was induced by both PI-3K-dependent and -independent pathways (11). Subsequent studies by Lin et al. (12), Shaw et al. (13), and Bijur et al. (7) have demonstrated that heat shock-induced activation of Akt can be completely inhibited by the PI-3K inhibitors LY294002 and wortmannin, demonstrating that heat shock-induced activation of Akt is PI-3K-dependent. Although heat shock robustly activates Akt, a corresponding decrease in GSK-3β activity has not been demonstrated, unlike the down-regulation of GSK-3β caused by growth factor stimulated Akt. Instead, He et al. (14) reported that transfer of heat shock-treated cells to control temperature resulted in a dramatic increase in GSK-3β activity by an unknown mechanism. Thus, it appears that, although growth factors and heat shock robustly activate Akt, the downstream regulations of GSK-3β by these two types of Akt activators appear to be quite distinct.
Although the mechanisms by which GSK-3β-facilitates apoptosis have not yet been identified, regulation of the activities of transcription factors may be involved. GSK-3β has been found to be an inhibitor of the heat shock factor-1 (HSF-1) transcription factor (14, 15). The ‘heat shock response’, as it is commonly known, is a protective mechanism that is rapidly activated in response to potentially lethal conditions, such as a rise in temperature, oxidative stress, osmotic stress, or other insults. Heat shock initiates a complex, incompletely understood, signaling pathway that activates the HSF-1 (16). Activation of HSF-1 increases the expression of heat shock proteins, such as hsp70 (17), which can protect cells against potentially lethal conditions. Activation of GSK-3β suppresses the transcriptional activity of HSF-1 through phosphorylation of Ser303 of HSF-1 (14, 15). Therefore, inadequate inhibitory control of GSK-3β activity is likely to impair the expression of heat shock proteins by inhibiting HSF-1 activation, and thus to increase cellular susceptibility to stress-induced death (7).
GSK-3β also can phosphorylate and down-regulate the activity of other transcription factors, such as AP-1, CREB, and Myc. GSK-3β functions as a negative regulator of the transcription factor AP-1 by phosphorylating c-Jun, one of the components of AP-1, resulting in a decrease in AP-1 DNA-binding activity (18). Troussard et al. (19) recently reported that inhibition of GSK-3β activity through phosphorylation by integrin-linked kinase could activate AP-1 activity, and result in regulation of cell survival, cell proliferation, cell differentiation, and cell migration. Similarly, reduction of CREB activity has been reported to facilitate apoptosis (20, 21), and one study found that GSK-3β functionally reduces CREB activity (22). Transcription factors Myc and Max form heterodimers that promote cell proliferation (23). Phosphorylation of Myc at Thr-58 or Ser-62 by GSK-3β was reported to negatively regulate Myc function, resulting in decreased cellular growth and transformation (24, 25). Thus, these and other studies indicate that the regulatory influence of GSK-3β on cell fate may be linked to its regulatory control of a variety of transcription factors.
In summary, over-activation, or inadequate inhibitory regulation, of GSK-3β can contribute to apoptosis. The activity of GSK-3β is normally negatively controlled by several cell survival signals, such as growth factor-induced activation of protein kinases upstream from GSK-3β. When the normal regulation of GSK-3β is impaired, such as when modeled by treatment with inhibitors of PI-3K, the uncontrolled activation of GSK-3β can initiate an array of changes in transcription factor activities that may contribute to the apoptotic process.
Lithium regulates GSK-3β
Lithium, one of the most effective drugs for the treatment of bipolar disorder, also has dramatic effects on morphogenesis in the early development of many organisms. This effect of lithium cannot be explained by its regulation of other signaling systems, such as inhibition of inositol phosphatases. In 1996, two groups reported that lithium inhibits GSK-3β, an enzyme that regulates cell fate determination in diverse organisms (26, 27). According to these studies, lithium directly and reversibly inhibits GSK-3β (26, 27). Although lithium may inhibit multiple enzymes, GSK-3β and GSK-3α are the only known protein kinases that are directly inhibited by lithium.
We have been interested in exploring the potential neuronal protective effects of lithium through inhibition of GSK-3β (7, 28). This effect of lithium was studied in human SH-SY5Y neuroblastoma cells transfected with GSK-3β. GSK-3β-overexpression in human SH-SY5Y neuroblastoma cells to a level that is 350% of the basal GSK-3β level does not cause spontaneous cell death, but increases the sensitivity of these cells to stress-induced apoptosis. Overexpression of GSK-3β increased apoptosis induced by both staurosporine and heat shock, and lithium treatment attenuated GSK-3β-facilitated apoptosis (7). These protective effects of lithium are illustrated in Fig. 1A, which shows that overexpression of GSK-3β-facilitated heat shock-induced apoptosis, as assessed by measurements of the activity of caspase-3, and that lithium treatment reduced the GSK-3β-facilitated component of apoptosis. In these experiments, protection by lithium required supra-therapeutic concentrations of lithium. That this requirement is due to the model system employed, GSK-3β-overexpressing cells, is illustrated in Fig. 1B. Based on the concentration-dependent inhibition by lithium of GSK-3β reported by Klein and Melton (26), we calculated the theoretical inhibition of overexpressed GSK-3β (350% of basal levels) in these cells. These results show that, because GSK-3β was overexpressed, 5 mM lithium is necessary just to reduce GSK-3β to the normal basal level, and higher lithium concentrations are necessary to reduce GSK-3β activity to below the normal basal level. These results indicate that reducing GSK-3β activity to below normal levels by lithium provides neuroprotection.
Fig. 1.
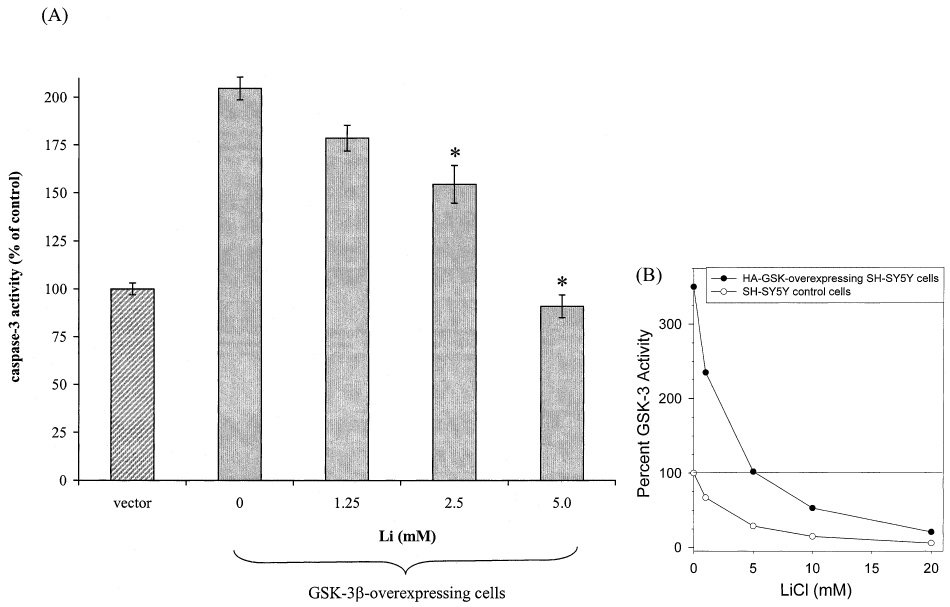
Lithium protects against heat shock-induced caspase-3 activation in GSK-3β-overexpressing SH-SY5Y cells. (A) Vector- and GSK-3β-transfected cells were pretreated with 0, 1.25, 2.5, or 5 mM lithium for 24 h, subjected to heat shock (45°C, 0.5 h), transferred to 37°C for 1 h, and caspase-3 activity was measured, as described previously (7). Values are expressed as the percentage of caspase-3 activity in vector-transfected cells (means ± SEM, n = 3. *p < 0.05 compared with values in GSK-3β-transfected cells not treated with lithium). (B) Data from Klein and Melton (26) showing the inhibition of GSK-3β by lithium were reproduced (open symbols) and used to calculate the theoretical activity of GSK-3β (filled symbols) in cells overexpressing GSK-3β (350% of basal GSK-3β activity).
In NTERA2 cells, lithium mimicked activation of the Wnt signaling pathway by increasing β-catenin levels, a response known to be induced by inhibition of GSK-3β (29). Because of the similarities between lithium’s actions and the Wnt signaling pathway, in that they both involve inhibition of GSK-3β, there has been interest in studying the Wnt pathway in connection with bipolar disorder. For example, Rhoads et al. (30) identified several genes in the Wnt pathway, such as Wnt-7a, that map within potential regions underlying susceptibility to bipolar disorder, schizophrenia, and disorders of neurodevelopmental origin. Thus, the finding that lithium selectively inhibits GSK-3β has led to many new areas for further research.
Another area of research that has received much attention is the regulation of cytoskeletal proteins by GSK-3β and the consequences of inhibition of GSK-3β by lithium. Treatment of cells with lithium inhibits the GSK-3β-dependent phosphorylation of the microtubule-associated protein tau (27, 31). This action may be important in regulating microtubule dynamics and cytoskeletal-based structural modifications associated with neuronal plasticity, effects that, in conjunction with changes in transcription factors, may contribute to the therapeutic actions of lithium.
Regulation of GSK-3β by anticonvulsant mood stabilizers
Clinically, anticonvulsant mood stabilizers have been established as an important addition to the classical mood stabilizer lithium in the treatment of bipolar disorder. Anticonvulsants have been reported to have a variety of effects on bipolar disorder. The efficacy of valproic acid (VPA) and of carbamazepine (CBZ) has been established for mania, and they may be more effective than lithium in rapid cycling and dysphoric mania (32). The establishment of VPA and CBZ as mood stabilizers stimulated the investigation and development of a new generation of anticonvulsants as potential mood stabilizers. For example, current data show that lamotrigine (LTG) has efficacy in bipolar depression (33, 34) and rapid cycling (35), but is less likely to be beneficial in mania.
The expanded use of anticonvulsants as mood stabilizers has raised the question of whether they have some of the same regulatory effects as lithium on intracellular signal transduction pathways. However, since this is a recent development, few investigations have addressed this topic. One of the most interesting findings was recently reported by Chen et al. (36), who found that VPA inhibits the activity of GSK-3β in human SH-SY5Y neuroblastoma cells. This mutual site of action of lithium and VPA raises the exciting possibility that inhibition of GSK-3β, or signaling systems associated with GSK-3β, may be important in the therapeutic effects of mood stabilizers.
Based on our findings that part of lithium’s neuroprotective properties results from its inhibition of GSK-3β, we hypothesized that anticonvulsant mood stabilizers also may suppress stress-induced cell death through inhibition of GSK-3β activity. To test this hypothesis, experiments were carried out in human SH-SY5Y neuroblastoma cells that overexpress active GSK-3β, as previously described (7). The potential antiapoptotic effects of anticonvulsant mood stabilizers were determined by measuring the activity of caspase-3, a cysteine protease which can be activated by apoptotic signals to initiate the cell death cascade.
Staurosporine-induced caspase-3 activity
A representative result showing staurosporine-induced activation of caspase-3 activity is shown in Fig. 2. Vector- or GSK-3β-transfected SH-SY5Y cells were treated with the indicated concentrations of staurosporine for 3 h. In vector-transfected cells, staurosporine only slightly increased caspase-3 activity (1.7-fold at 0.05 µM, and 3.5-fold at the higher concentrations). In contrast, staurosporine treatment caused a very large increase in caspase-3 activity in GSK-3β-transfected cells, causing a 17-fold increase at 0.05 µM, a 32-fold increase at 0.1 µM, and a 61-fold increase at 0.5 µM staurosporine.
Fig. 2.
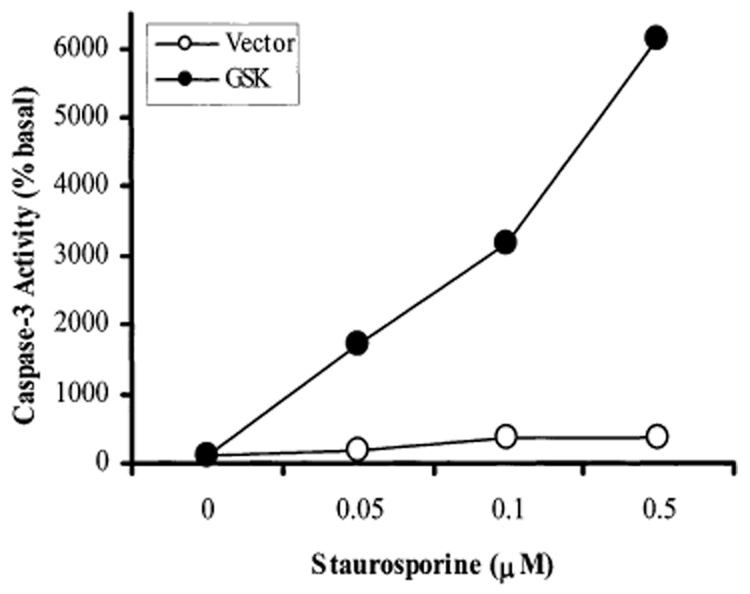
Staurosporine-stimulated caspase-3 activity in vector-transfected and GSK-3β-transfected SH-SY5Y cells. Vector-transfected cells (open circles) or GSK-3β-transfected cells (solid circles) were treated with 0, 0.05, 0.1, and 0.5 µM staurosporine for 3 h. Cells were harvested and caspase-3 activity in cell lysates was measured with DEVD-AMC, a caspase-3 substrate, as described previously (7). Values are percentages of caspase-3 activity in untreated cells (no staurosporine). A representative of three experiments is shown.
Using GSK-3β-transfected SH-SY5Y cells, we tested the effects of the anticonvulsant mood stabilizers, VPA, CBZ, and LTG, on staurosporine-induced caspase-3 activity (Fig. 3 and Fig. 4). VPA did not cause a significant change in the basal caspase-3 activity. However, staurosporine (0.5 µM)-stimulated caspase-3 activity was inhibited by 40% after the GSK-3β-transfected cells were treated with VPA at a therapeutically relevant concentration (0.6 mM) for 7 days (Fig. 3A). This result is consistent with the report of Chen et al. (36) that VPA can inhibit the activity of GSK-3β. In contrast, a therapeutic concentration of CBZ (0.05 mM) did not inhibit staurosporine-increased caspase-3 activity in GSK-3β-transfected cells (Fig. 3B). LTG at 0.1 mM concentration did not inhibit staurosporine-stimulated caspase-3 activity, and higher but therapeutic relevant concentrations of LTG (0.2 and 0.3 mM) significantly inhibited staurosporine (0.1 µM)-stimulated caspase-3 activity in GSK-3β-transfected cells (Fig. 4).
Fig. 3.
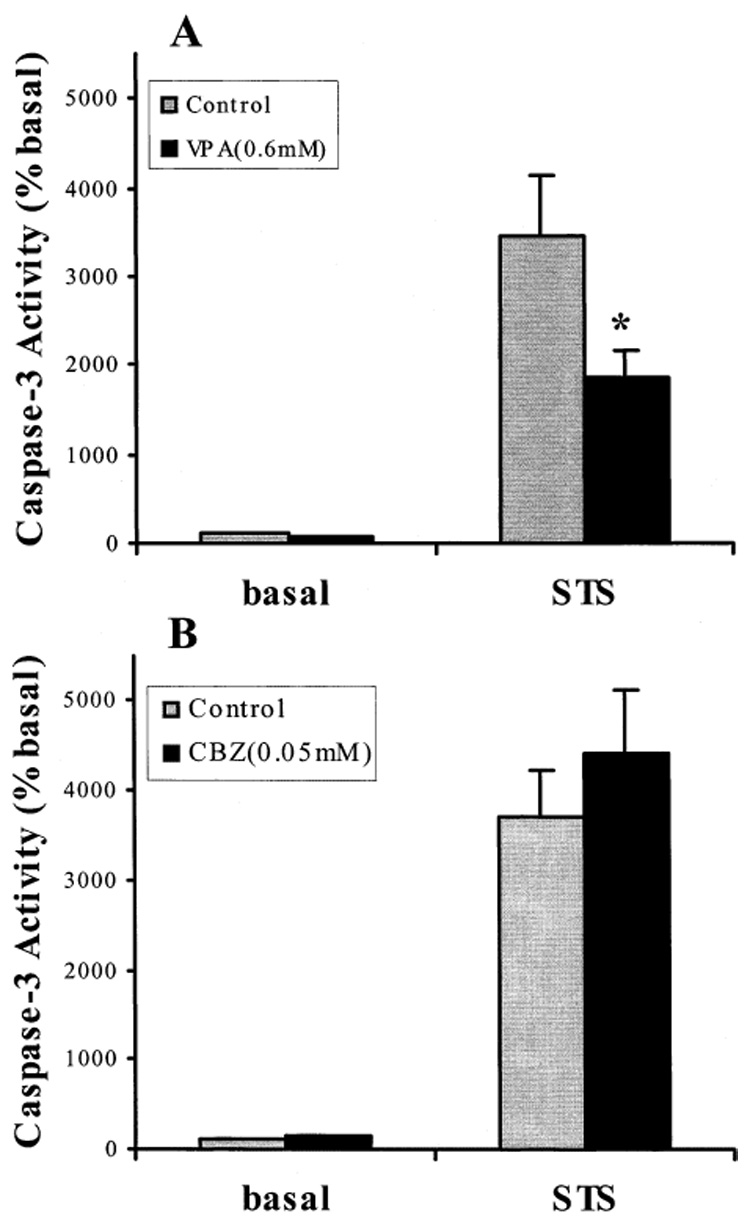
Effects of VPA and CBZ on staurosporine-stimulated caspase-3 activity in GSK-3β-transfected SH-SY5Y cells. Cells were not treated (control) or treated with 0.6 mM VPA (A) or with 0.05 mM CBZ (B) for 7 days, and then either not further treated (basal) or treated with 0.5 µM staurosporine (STS) for 3 h. Cells were harvested and caspase-3 activity was measured as described previously (7). Values are percentages of the caspase-3 activity in untreated cells (means ± SEM, n = 10, *p < 0.05 comparing VPA or CBZ treatment to control).
Fig. 4.
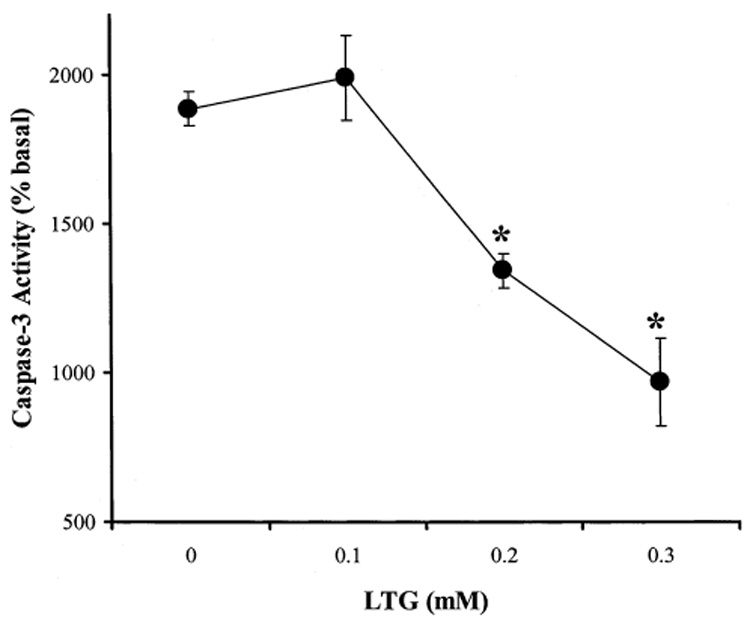
Effect of LTG on staurosporine-stimulated caspase-3 activity in GSK-3β-transfected SH-SY5Y cells. Cells were not treated (0) or treated with 0.1, 0.2, or 0.3 mM LTG for 7 days. Cells were then treated with 0.1 µM staurosporine for 3 h. Cells were harvested and caspase-3 activity was measured as described previously (7). Values are percentages of the caspase-3 activity in untreated cells (means ± SEM, n = 3, *p < 0.05 comparing LTG treatment to control).
Heat shock-induced caspase-3 activity
As with staurosporine, heat shock only mildly increased caspase-3 activity in vector-transfected SH-SY5Y cells, causing a two-fold increase after 30 min at 45°C followed by a 30 min recovery incubation at 37°C, and four-fold increase after a 90 min recovery incubation (Fig. 5A). In GSK-3β-transfected cells, caspase-3 activity continued to increase up to 18-fold when post-heat shock recovery was extended to 90 min. VPA (0.6 mM) significantly inhibited heat shock-induced caspase-3 activity after recovery for either 30 or 90 min (Fig. 4A). The effects of different anticonvulsants in heat shock-induced caspase-3 activity were compared, as shown in Fig. 5B. Chronic (7 days) treatment of GSK-3β-transfected SH-SY5Y cells with therapeutically relevant concentrations of VPA (0.6 mM), CBZ (0.05 mM), and LTG (0.1 and 0.3 mM) elicited divergent effects. VPA caused 50% inhibition, CBZ had no effect, 0.1 mM LTG caused slight inhibition, and 0.3 mM LTG caused about 55% inhibition of heat shock-induced caspase-3 activity.
Fig. 5.
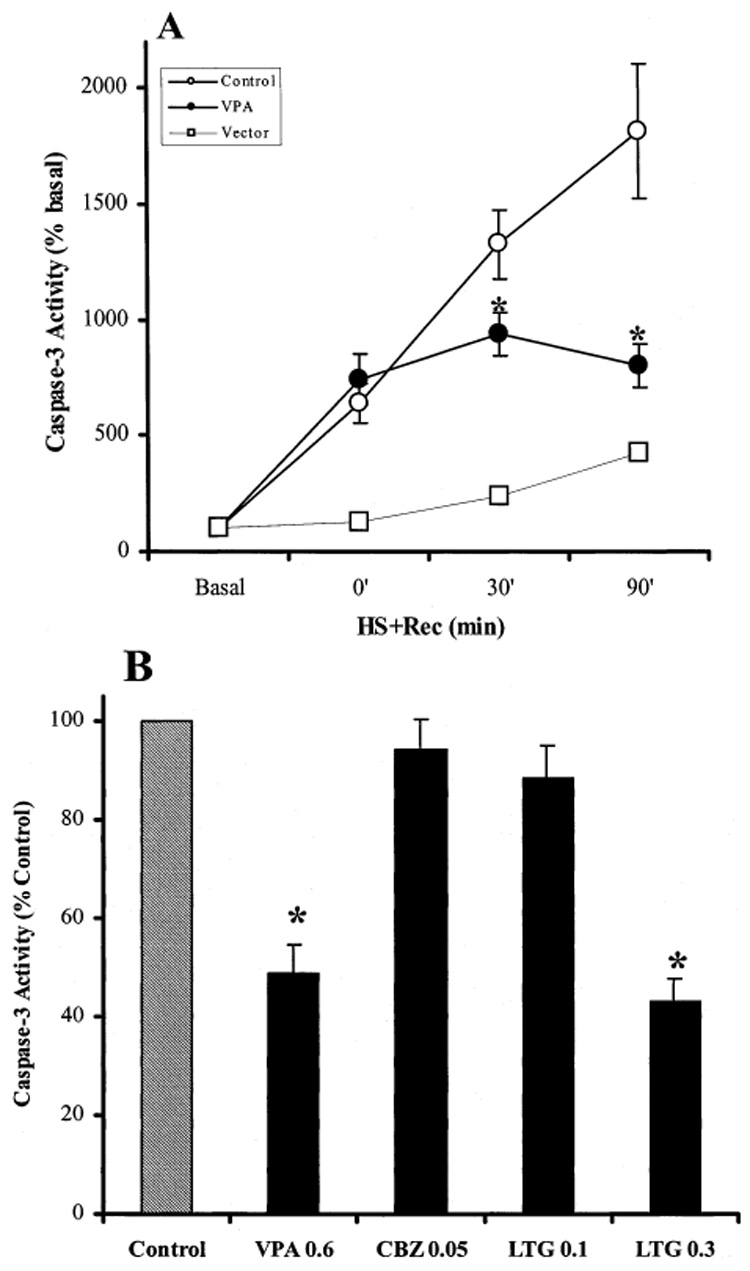
Effects of anticonvulsants on heat shock-induced caspase-3 activity in GSK-3β-transfected SH-SY5Y cells. (A) Vector transfected cells (open squares) or GSK-3β-transfected cells (circles) were exposed to heat shock at 45°C for 30 min and then incubated at 37°C (HS + Rec) for 0, 30, and 90 min. Cells were treated without (open symbols) or with (closed symbols) 0.6 mM VPA for 7 days prior to heat shock treatment. In vector transfected cells, a representative of three experiments is shown. In GSK-3β-transfected cells, values are percentage of the caspase-3 activity in untreated cells. (means ± SEM, n = 8–11, *p < 0.05 compared to cells without treatment). (B) GSK-3β-transfected SH-SY5Y cells were treated with 0.6 mM VPA, 0.05 mM CBZ, or 0.1 and 0.3 mM LTG for 7 days before exposure to heat shock for 30 min with a 90 min recovery incubation at 37°C. Values are percentage of control (heat shock without anticonvulsant treatment). (means ± SEM, n = 9, *p < 0.05 compared to cells with no anticonvulsant treatment).
In summary, chronic VPA at a therapeutically relevant concentration significantly inhibited staurosporine- and heat shock-induced apoptosis facilitated by GSK-3β, as measured by changes in caspase-3 activity. CBZ, in contrast, had no effect on either staurosporine- or heat shock-stimulated caspase-3 activity in GSK-3β-overexpressing cells, although it remains to be investigated if CBZ regulates signals downstream of GSK-3β. Interestingly, chronic treatment with 0.2–0.3 mM LTG significantly inhibited staurosporine- and heat shock-stimulated caspase-3 activity in GSK-3β-overexpressing cells, an effect similar to lithium and VPA. This is the first finding indicating that LTG may regulate intracellular signal transduction systems and provide neuroprotection through regulation of GSK-3β-associated signaling pathways. Therefore, these results indicate that the anticonvulsant mood stabilizers VPA and LTG may have neuroprotective actions similar to lithium. Whether these effects contribute to the anti-bipolar properties of VPA and LTG remains to be identified.
Conclusion
It has recently become clear that GSK-3β facilitates apoptosis. In normal cells, GSK-3β activity is under the inhibitory control of growth factors through a series of kinases upstream from GSK-3β. Dysregulation of GSK-3β, resulting in increased activity, facilitates apoptosis, perhaps in part through GSK-3β’s inhibitory effects on transcription factors, such as HSF-1, CREB, and others, that normally contribute to cell survival.
Lithium is a selective inhibitor of GSK-3β, and this action of lithium may contribute in part to its neuroprotective function. By establishing a model system of cells overexpressing GSK-3β, we have established conditions in which a specific component of apoptosis can be attributed directly to hyperactive GSK-3β, thus protection from the GSK-3β component of apoptosis directly reveals a neuroprotective mechanism of action of effective drugs. In other words, the GSK-3β-dependent component of apoptosis has been experimentally isolated, allowing determinations of whether drugs provide protection specifically from the GSK-3β-dependent component of apoptosis. Thus, rather than relying on tenuous correlations between events (e.g., cell survival and expression of a particular gene), a cause-and-effect experimental situation has been established; GSK-3β causes the facilitated apoptosis, do the drugs protect from GSK-3β facilitated apoptosis? In addition to lithium, two out of the three anticonvulsant mood stabilizers that were tested had the ability to attenuate apoptosis mediated by GSK-3β hyperactivity. These findings indicate that mood stabilizers can have neuroprotective functions associated with signaling systems involving GSK-3β. These findings also add to an extensive literature that has demonstrated the neuroprotective effects of lithium (e.g., (37–41)), and some reported evidence for neuroprotection by VPA (42, 43), and LTG (44–46), by identifying a specific mechanism contributing to their neuroprotective effects, the attenuation of signaling systems associated with GSK-3β. These may include the signaling pathways that regulate the activity of GSK-3β and/or the substrate proteins that are regulated by GSK-3β. For example, these drugs may activate signals that contribute to the inhibition of GSK-3β, such as the PI3k/Akt pathway, they may directly inhibit GSK-3β, notably as do lithium and VPA, or they may contribute to the regulation of substrates of GSK-3β and downstream consequences, such as activation of HSF-1, which is normally inhibited by GSK-3β and/or facilitation of the expression of heat shock proteins that are regulated by HSF-1. Although the pathophysiology of bipolar disorder is unknown, the finding that three out of four established or promising mood stabilizers have inhibitory effects on GSK-3β-mediated signaling pathways suggests that abnormal GSK-3β expression or activity, or signaling systems connected to GSK-3β, may contribute to the pathophysiology of bipolar disorder.
Acknowledgements
This research was supported by NIH grant MH38752.
References
- 1.Jope RS. Anti-bipolar therapy: mechanism of action of lithium. Molec Psychiatry. 1999;4:117–128. doi: 10.1038/sj.mp.4000494. [DOI] [PubMed] [Google Scholar]
- 2.Hartigan JA, Johnson GV. Transient increases in intracellular calcium result in prolonged site-selective increases in Tau phosphorylation through a glycogen synthase kinase-3beta-dependent pathway. J Biol Chem. 1999;274:21395–21401. doi: 10.1074/jbc.274.30.21395. [DOI] [PubMed] [Google Scholar]
- 3.Cook D, Fry MJ, Hughes K, Sumathipala R, Woodgett JR, Dale TC. Wingless inactivates glycogen synthase kinase-3 via an intracellular signalling pathway which involves a protein kinase C. EMBO J. 1996;15:4526–4536. [PMC free article] [PubMed] [Google Scholar]
- 4.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 5.Takashima A, Noguchi K, Sato K, Hoshino T, Imahori K. Tau protein kinase I is essential for amyloid beta-protein-induced neurotoxicity. Pro Natl Acad Sci USA. 1993;90:7789–7793. doi: 10.1073/pnas.90.16.7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pap M, Cooper GM. Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-Kinase/Akt cell survival pathway. J Biol Chem. 1998;273:19929–19932. doi: 10.1074/jbc.273.32.19929. [DOI] [PubMed] [Google Scholar]
- 7.Bijur GN, De Sarno P, Jope RS. Glycogen synthase kinase-3beta facilitates staurosporine- and heat shock-induced apoptosis. Protection by lithium. J Biol Chem. 2000;275:7583–7590. doi: 10.1074/jbc.275.11.7583. [DOI] [PubMed] [Google Scholar]
- 8.Hetman M, Cavanaugh JE, Kimelman D, Xia Z. Role of glycogen synthase kinase-3beta in neuronal apoptosis induced by trophic withdrawal. J Neuroscience. 2000;20:2567–2574. doi: 10.1523/JNEUROSCI.20-07-02567.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan TO, Rittenhouse SE, Tsichlis PN. AKT/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Ann Rev Biochem. 1999;68:965–1014. doi: 10.1146/annurev.biochem.68.1.965. [DOI] [PubMed] [Google Scholar]
- 10.Konishi H, Matsuzaki H, Tanaka M, Takemura Y, Kuroda S, Ono Y, Kikkawa U. Activation of protein kinase B (Akt/RAC-protein kinase) by cellular stress and its association with heat shock protein Hsp27. FEBS Letters. 1997;410:493–498. doi: 10.1016/s0014-5793(97)00541-3. [DOI] [PubMed] [Google Scholar]
- 11.Konishi H, Fujiyoshi T, Fukui Y, Matsuzaki H, Yamamoto T, Ono Y, Andjelkovic M, Hemmings BA, Kikkawa U. Activation of protein kinase B induced by H(2)O(2) and heat shock through distinct mechanisms dependent and independent of phosphatidylinositol 3-kinase. J Biochem. 1999;126:1136–1143. doi: 10.1093/oxfordjournals.jbchem.a022559. [DOI] [PubMed] [Google Scholar]
- 12.Lin RZ, Hu ZW, Chin JH, Hoffman BB. Heat shock activates c-Src tyrosine kinases and phosphatidylinositol 3-kinase in NIH3T3 fibroblasts. J Biol Chem. 1997;272:31196–31202. doi: 10.1074/jbc.272.49.31196. [DOI] [PubMed] [Google Scholar]
- 13.Shaw M, Cohen P, Alessi DR. The activation of protein kinase B by H2O2 or heat shock is mediated by phosphoinositide 3-kinase and not by mitogen-activated protein kinase-activated protein kinase-2. Biochem J. 1998;336:241–246. doi: 10.1042/bj3360241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He B, Meng YH, Mivechi NF. Glycogen synthase kinase-3beta and extracellular signal-regulated kinase inactivate heat shock transcription factor 1 by facilitating the disappearance of transcriptionally active granules after heat shock. Molec Cell Biol. 1998;18:6624–6633. doi: 10.1128/mcb.18.11.6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chu B, Soncin F, Price BD, Stevenson MA, Calderwood SK. Sequential phosphorylation by mitogen-activated protein kinase and glycogen synthase kinase-3 represses transcriptional activation by heat shock factor-1. J Biol Chem. 1996;271:30847–30857. doi: 10.1074/jbc.271.48.30847. [DOI] [PubMed] [Google Scholar]
- 16.Morimoto RI. Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998;12:3788–3796. doi: 10.1101/gad.12.24.3788. [DOI] [PubMed] [Google Scholar]
- 17.Feder ME, Hofmann GE. Heat-shock proteins, molecular chaperones, and the stress response: evolutionary and ecological physiology. Ann Rev Physiol. 1999;61:243–282. doi: 10.1146/annurev.physiol.61.1.243. [DOI] [PubMed] [Google Scholar]
- 18.Boyle WJ, Smeal T, Defize LH, Angel P, Woodgett JR, Karin M, Hunter T. Activation of protein kinase C decreases phosphorylation of c-Jun at sites that negatively regulate its DNA-binding activity. Cell. 1991;64:573–584. doi: 10.1016/0092-8674(91)90241-p. [DOI] [PubMed] [Google Scholar]
- 19.Troussard AA, Tan C, Yoganathan TN, Dedhar S. Cell-extracellular matrix interactions stimulate the AP-1 transcription factor in an integrin-linked kinase- and glycogen synthase kinase-3-dependent manner. Molec Cell Biol. 1999;19:7420–7427. doi: 10.1128/mcb.19.11.7420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jean D, Harbison M, McConkey DJ, Ronai Z, Bar-Eli M. CREB and its associated proteins act as survival factors for human melanoma cells. J Biol Chem. 1998;273:24884–24890. doi: 10.1074/jbc.273.38.24884. [DOI] [PubMed] [Google Scholar]
- 21.Walton M, Woodgate AM, Muravlev A, Xu R, During MJ, Dragunow M. CREB phosphorylation promotes nerve cell survival. J Neurochem. 1999;73:1836–1842. [PubMed] [Google Scholar]
- 22.Bullock BP, Habener JF. Phosphorylation of the cAMP response element binding protein CREB by cAMP-dependent protein kinase A and glycogen synthase kinase-3 alters DNA-binding affinity, conformation, and increases net charge. Biochemistry. 1998;37:3795–3809. doi: 10.1021/bi970982t. [DOI] [PubMed] [Google Scholar]
- 23.Amati B, Land H. Myc-Max-Mad: a transcription factor network controlling cell cycle progression, differentiation and death. Curr Opin Gen Dev. 1994;4:102–108. doi: 10.1016/0959-437x(94)90098-1. [DOI] [PubMed] [Google Scholar]
- 24.Henriksson M, Bakardjiev A, Klein G, Luscher B. Phosphorylation sites mapping in the N-terminal domain of c-myc modulate its transforming potential. Oncogene. 1993;8:3199–3209. [PubMed] [Google Scholar]
- 25.Pulverer BJ, Fisher C, Vousden K, Littlewood T, Evan G, Woodgett JR. Site-specific modulation of c-Myc cotransformation by residues phosphorylated in vivo. Oncogene. 1994;9:59–70. [PubMed] [Google Scholar]
- 26.Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Pro Natl Acad Sci USA. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stambolic V, Ruel L, Woodgett JR. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr Biol. 1997;6:1664–1668. doi: 10.1016/s0960-9822(02)70790-2. [DOI] [PubMed] [Google Scholar]
- 28.Bijur GN, Jope RS. Opposing actions of phosphatidylinositol 3-kinase and GSK-3beta in the regulation of HSF-1 activity. J Neurochem. 2000;75:2401–2408. doi: 10.1046/j.1471-4159.2000.0752401.x. [DOI] [PubMed] [Google Scholar]
- 29.Giesberts AN, Duran C, Morton IN, Pigott C, White SJ, Andrews PW. The expression and function of cadherin-mediated cell-to-cell adhesion in human embryonal carcinoma cells. Mech Dev. 1999;83:115–125. doi: 10.1016/s0925-4773(99)00043-x. [DOI] [PubMed] [Google Scholar]
- 30.Rhoads AR, Karkera JD, Detera-Wadleigh SD. Radiation hybrid mapping of genes in the lithium-sensitive wnt signaling pathway. Molec Psychiatry. 1999;4:437–442. doi: 10.1038/sj.mp.4000538. [DOI] [PubMed] [Google Scholar]
- 31.Takahashi M, Yasutake K, Tomizawa K. Lithium inhibits neurite growth and tau protein kinase I/glycogen synthase kinase-3beta-dependent phosphorylation of juvenile tau in cultured hippocampal neurons. J Neurochem. 1999;73:2073–2083. [PubMed] [Google Scholar]
- 32.Post RM, Denicoff KD, Frye MA, Dunn RT, Leverich GS, Osuch E, Speer A. A history of the use of anticonvulsants as mood stabilizers in the last two decades of the 20th century. Neuropsychobiology. 1998;38:152–166. doi: 10.1159/000026532. [DOI] [PubMed] [Google Scholar]
- 33.Calabrese JR, Bowden CL, Sachs GS, Ascher JA, Monaghan E, Rudd GD. A double-blind placebo-controlled study of lamotrigine monotherapy in outpatients with bipolar I depression. Lamictal 602 Study Group. J Clin Psychiatry. 1999;60:79–88. doi: 10.4088/jcp.v60n0203. [DOI] [PubMed] [Google Scholar]
- 34.Frye MA, Ketter TA, Kimbrell TA, Dunn RT, Speer AM, Osuch EA, Luckenbaugh DL, Cora-Locatelli G, Leverich G, Post RM. A placebo controlled study of lamotrigine and gabapentin monotherpay in refractory mood disorders. J Clin Psychopharmacol. 2000;20:607–614. doi: 10.1097/00004714-200012000-00004. [DOI] [PubMed] [Google Scholar]
- 35.Bowden CL, Calabrese JR, McElroy SL, Rhodes LJ, Keck PE, Jr, Cookson J, Anderson J, Bolden-Watson C, Ascher J, Monaghan E, Zhou J. The efficacy of lamotrigine in rapid cycling and non-rapid cycling patients with bipolar disorder. Biol Psychiatry. 1999;45:953–958. doi: 10.1016/s0006-3223(99)00013-x. [DOI] [PubMed] [Google Scholar]
- 36.Chen G, Huang LD, Jiang YM, Manji HK. The mood-stabilizing agent valproate inhibits the activity of glycogen synthase kinase-3. J Neurochem. 1999;72:1327–1330. doi: 10.1046/j.1471-4159.2000.0721327.x. [DOI] [PubMed] [Google Scholar]
- 37.Volonte C, Rukenstein A. Lithium chloride promotes short-term survival of PC12 cells after serum and NGF deprivation. Lithium. 1993;4:211–219. [Google Scholar]
- 38.Nonaka S, Hough CJ, Chuang DM. Chronic lithium treatment robustly protects neurons in the central nervous system against excitotoxicity by inhibiting N-methyl-d-aspartate receptor-mediated calcium influx. Proc Natl Acad Sci USA. 1998;95:2642–2647. doi: 10.1073/pnas.95.5.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nonaka S, Chuang DM. Neuroprotective effects of chronic lithium on focal cerebral ischemia in rats. Neuroreport. 1998;9:2081–2084. doi: 10.1097/00001756-199806220-00031. [DOI] [PubMed] [Google Scholar]
- 40.Centeno F, Mora A, Fuentes JM, Soler G, Claro E. Partial lithium-associated protection against apoptosis induced by C2-ceramide in cerebellar granule neurons. Neuroreport. 1989;9:4199–4203. doi: 10.1097/00001756-199812210-00036. [DOI] [PubMed] [Google Scholar]
- 41.Wei H, Leeds PR, Qian Y, Wei W, Chen R, Chuang D. β-amyloid peptide-induced death of PC 12 cells and cerebellar granule cell neurons is inhibited by long-term lithium treatment. Eur J Pharmacol. 2000;392:117–123. doi: 10.1016/s0014-2999(00)00127-8. [DOI] [PubMed] [Google Scholar]
- 42.Mora A, Gonzalez-Polo RA, Fuentes JM, Soler G, Centeno F. Different mechanisms of protection against apoptosis by valproate and Li+ Eur J Biochem. 1999;266:886–8891. doi: 10.1046/j.1432-1327.1999.00919.x. [DOI] [PubMed] [Google Scholar]
- 43.Bruno V, Sortino MA, Scapagnini U, Nicoletti F, Canonico PL. Antidegenerative effects of Mg(2+)-valproate in cultured cerebellar neurons. Funct Neurol. 1995;10:121–130. [PubMed] [Google Scholar]
- 44.Wiard RP, Dickerson MC, Beek O, Norton R, Cooper BR. Neuroprotective properties of the novel antiepileptic lamotrigine in a gerbil model of global cerebral ischemia. Stroke. 1995;26:466–472. doi: 10.1161/01.str.26.3.466. [DOI] [PubMed] [Google Scholar]
- 45.Casanovas A, Ribera J, Hukkanen M, Riveros-Moreno V, Esquerda JE. Prevention by lamotrigine, MK-801 and N omega-nitro-l-arginine methyl ester of motoneuron cell death after neonatal axotomy. Neuroscience. 1996;71:313–325. doi: 10.1016/0306-4522(95)00461-0. [DOI] [PubMed] [Google Scholar]
- 46.Connop BP, Boegman RJ, Beninger RJ, Jhamandas K. Malonate-induced degeneration of basal forebrain cholinergic neurons:attenuation by lamotrigine, MK-801, and 7-nitroindazole. J Neurochem. 1997;68:1191–1199. doi: 10.1046/j.1471-4159.1997.68031191.x. [DOI] [PubMed] [Google Scholar]