

Abstract
Adoptive cell therapy (ACT) using autologous tumour-infiltrating lymphocytes has emerged as the most effective treatment for patients with metastatic melanoma and can mediate objective cancer regression in approximately 50% of patients. The use of donor lymphocytes for ACT is an effective treatment for immunosuppressed patients who develop post-transplant lymphomas. The ability to genetically engineer human lymphocytes and use them to mediate cancer regression in patients, which has recently been demonstrated, has opened possibilities for the extension of ACT immunotherapy to patients with a wide variety of cancer types and is a promising new approach to cancer treatment.
Adoptive cell therapy (ACT) has emerged as the most effective treatment for patients with metastatic melanoma. ACT-based immunotherapy was first described in 1988 (REF. 1), but the decisive improvement in efficacy came in 2002 with the introduction of an immunodepleting preparative regimen given before the adoptive transfer, which could result in the clonal repopulation of patients with anti-tumour T cells2. Of patients with metastatic melanoma refractory to all other treatments, 50% will experience an objective response, some with complete responses3. Responses can be durable and are seen in all organ sites, including the brain. Recent studies demonstrating that normal human lymphocytes can be genetically engineered to recognize cancer antigens and mediate cancer regression in vivo has opened opportunities for enhancing and extending the ACT approach to patients with a wide variety of cancer types4. These studies provide a valuable guide to the immunological principles that form the basis of effective immunotherapies for patients with cancer.
The role of ACT in human cancer immuotherapy
Current efforts in the immunotherapy of human solid cancers fall into three main categories.
Non-specific immunomodulation
This is mediated by the administration of the T-cell growth factor interleukin 2 (IL2) and can activate endogenous tumour-reactive cells in vivo and reproducibly cause the regression of some human solid cancers5–7. The durability of the cancer regressions induced by IL2 led to its approval by the US Food and Drug Administration for the treatment of patients with metastatic renal cancer in 1992 and metastatic melanoma in 1998. Although IL2 administration leads to toxicity owing to a capillary leak syndrome, experience with the administration of this cytokine has resulted in treatment-related mortalities of <1%8. More recently, antibody-mediated blockade of a cell surface inhibitory molecule, cytotoxic T-lymphocyte-associated 4 (CTLA4), has resulted in objective clinical responses in 10–20% of patients, but again only consistently in those with metastatic melanoma or renal cancer, suggesting that these two tumour types are exceptional in their ability to naturally generate endogenous anti-tumour cells of sufficient avidity and in sufficient numbers to mediate cancer regression when appropriately stimulated in vivo9,10. Investigations are underway to evaluate other general immune modulators such IL15, anti-transforming growth factor-β (anti-TGFβ) and anti-programmed death 1 (anti-PD-1) antibodies.
Active immunization approaches (cancer vaccines)
These are based on immunizing cancer patients against their autologous cancers using either whole cells, proteins, peptides or a wide variety of immunizing vectors. The identification of a large number of human cancer antigens beginning in 1991 fuelled a resurgence of interest in this area11,12. Currently, only rare and highly sporadic regressions of solid cancers have been achieved using active immunization13. Several recent findings have further tempered enthusiasm for this approach. Even though up to 30% of circulating anti-melanoma CD8+ T cells could be induced by immunization of patients with melanoma, tumour progression can occur, suggesting that the cells induced are of low avidity and/or subject to inhibition by endogenous factors14. Although increases in anti-tumour T cells have been suggested in some active immunization protocols in patients with cancers other than melanoma, it has been possible to isolate and grow only rare anti-tumour T cells from these tumour types, again suggesting that T-cell precursors reactive with non-melanoma antigens are present at low frequency13.
At a glance
Adoptive cell therapy (ACT) is a treatment that uses a cancer patient’s own T lymphocytes with anti-tumour activity, expanded in vitro and reinfused into the patient with cancer.
ACT using autologous tumour-infiltrating lymphocytes is currently the most effective treatment for patients with metastatic melanoma and can mediate objective tumour regressions in 50% of patients.
Lymphodepletion before ACT is an important component of the treatment because it eliminates T regulatory cells and eliminates lymphocytes, which compete with the transferred cells for homeostatic cytokines such as interleukin 7 (IL7) and IL15.
ACT can be effective in treating selected patients with post-transplant lymphoproliferative diseases (PTLD) resulting from Epstein–Barr virus, which can cause PTLD during the immunosuppressed state.
Recent studies have shown that genetic modification of lymphocytes using retroviruses that encode T-cell receptors can convert normal lymphocytes into lymphocytes with anti-cancer activity. The adoptive transfer of these lymphocytes into patients with metastatic melanoma can mediate tumour regression.
ACT
This approach involves the identification ex vivo of autologous or allogeneic lymphocytes with antitumour activity, which are then infused into cancer patients, often along with appropriate growth factors to stimulate their survival and expansion in vivo. ACT has substantial theoretical and practical advantages over the approaches discussed above. It is necessary to identify only a small number of anti-tumour cells with the appropriate properties that can then be expanded to large numbers ex vivo for treatment. In vitro tests can identify the exact populations and effector functions required for cancer regression, which can then be selected for expansion. The cells can be activated in the laboratory free from endogenous inhibitory factors and thus can be induced to exhibit the required anti-tumour effector functions. Perhaps most importantly, it is possible to manipulate the host before cell transfer to provide an optimal environment for the transferred cells. This approach has proved to be highly effective for the treatment of cancer in experimental animals as well as in cancer patients.
Adoptive cell transfer in animal models
Shortly after the demonstration that the cellular arm of the immune system was responsible for tissue rejection15, attempts were made to treat established rodent tumours by the transfer of immune cells. Examples of the development of ACT in animal models are summarized in TIMELINE 1. As T lymphocytes could not be grown in vitro, early efforts at ACT in the 1960s were limited to the use of cells obtained directly from immunized animals. Studies by Alexander and colleagues in the mid-1960s showed that sarcomas a few millimetres in size could be treated in rats by the administration of large numbers of lymphocytes from immunized syngeneic animals16. Extensive studies by Fefer and colleagues beginning in 1969 showed that intraperitoneal instillation of immune lymphocytes along with chemotherapy could effectively treat mice bearing intraperitoneal virus-induced lymphomas17. The ability to expand populations of anti-tumour immune cells using in vitro sensitization techniques in the mid-1970s freed these studies from the constraints imposed by the need for fresh cells from immunized hosts18. Eberlein et al. took this a step further by showing that the intravenous injection of immune cells grown in culture in IL2 could treat disseminated tumours in mice19 and subsequent studies showed that the concurrent administration of IL2 could further enhance the effectiveness of these IL2-dependent cells in vivo20.
Timeline 1. Selected highlights in the development of ACT in animal models.
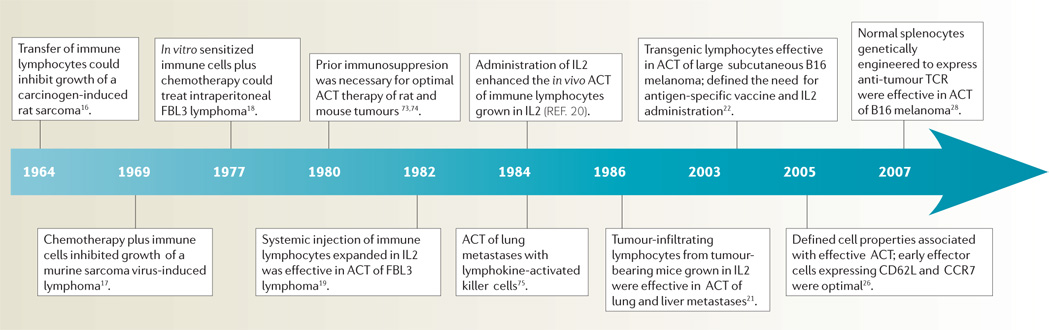
ACT, adoptive cell therapy; IL2, interleukin 2; TCR, T-cell receptor.
The need for immunization of lymphocyte donors limited the application of this approach until 1986 when it was shown that tumour-infiltrating lymphocytes (TIL) from non-immunized mice bearing sarcomas or melanomas could be expanded in vitro in IL2 and used to successfully treat established lung and liver tumours21.
More recently, T-cell receptor (TCR) transgenic mice, all of whose lymphocytes express anti-gp100 tumour antigen TCRs, have provided a constant source anti-tumour T cells that are valuable for defining host factors and cell properties associated with effective ACT of large, vascularized, subcutaneous tumours in mice22. The importance of host immunosuppression as part of ACT was shown to be due to both the elimination of T regulatory cells23 as well as the elimination of ‘cytokine sinks’ that compete with the transferred cells for homeostatic cytokines, such as IL7 and IL15, that are produced by host stromal cells24. The greater the degree of host lymphodepletion the more effective was the treatment25. The characteristics of the transferred cells themselves had a profound effect: anti-tumour T cells with a CCR7+,CD27+,CD28+,CD62L+ phenotype that is characteristic of central memory cells were more effective than highly differentiated cells that lost these markers26. Immunization of the host with a vaccine expressing a tumour antigen recognized by the transferred cells enhanced their therapeutic effect22. Antigen-presenting cells (APCs) also have a role in effective ACT as the injection of APC-stimulating molecules such as Toll-like receptor agonists enhanced the efficacy of treatment27. More recently, it was demonstrated that normal murine splenocytes transduced with a retrovirus encoding an anti-melanoma TCR resulted in the generation lymphocytes that recognized the melanoma in vitro and, when adoptively transferred, could mediate tumour regression in vivo28.
ACT is currently the most effective immunotherapy capable of mediating the rejection of large tumours in mice and optimization of the treatment in mouse models has had an important role in the design of ACT approaches in the human.
ACT in metastatic melanoma
Selected highlights in the development of ACT in patients with cancer are shown in TIMELINE 2.
Timeline 2. Examples of ACT in patients with cancer.
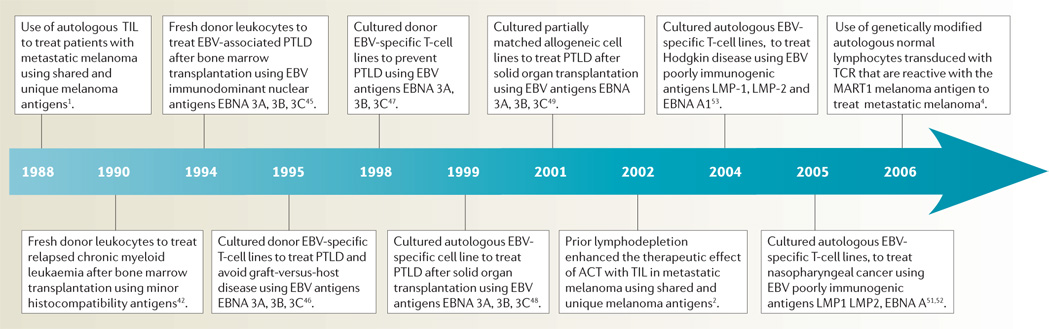
EBV, Epstein–Barr virus; PTLD, post-transplant lymphoproliferative disease; TCR, T-cell receptor; TIL, tumour-infiltrating lymphocytes.
An important step in the development of human ACT was the finding in 1987 that lymphocytes infiltrating melanomas could be grown in IL2 and exhibit major histocompatibility complex (MHC)-restricted recognition of the autologous melanoma29. Over 50 different antigenic epitopes either unique to the autologous tumour or widely shared among melanomas were characterized using these TIL12. Studies using improved culture methods capable of generating up to 1011 TIL showed that melanoma-specific activity could be detected in TIL from 81% of 36 consecutive patients30.
The infusion of autologous TIL grown from the resected tumour nodules of patients with metastatic melanoma represents the clearest example of the effectiveness of ACT for the treatment of patients with a metastatic solid cancer and has helped to elucidate the cellular and host characteristics required for effective treatment in humans. The original reports of this approach, first published in 1988 (REF. 1) and summarized in 1994 (REF. 31), described an overall objective response rate of 34% in 86 patients treated with autologous TIL plus high-dose IL2 (REF 31). Patients received two cycles of treatment separated by 2 weeks. Eighty-eight percent of patients received >1011 cells in the first cycle. Response rates were similar in patients treated alone (31%) or with low-dose cyclophosphamide (35%) and in patients who had not received prior IL2 (34%) or in patients refractory to prior IL2 therapy (32%). In this trial, TIL were administered regardless of their in vitro activity, although a retrospective analysis revealed that there was a highly significant correlation between clinical response and the ability of the TIL to lyse the autologous fresh tumour (P = 0.0008)32,33. Traffic of indium-111-labelled TIL to tumour deposits correlated with clinical response (P = 0.022). Shorter times in culture and shorter doubling times were also positively associated with response (P = 0.0001 and 0.03)33, in accord with the findings in mouse models that increased proliferative potential was an important property of clinically active cells.
Several shortcomings became apparent in these initial studies of ACT using TIL. Persistence of the transferred cells in vivo was short. Studies using retroviral insertion of the neomycin phosphotransferase gene to mark TIL and sensitive PCR assays to detect the transgene revealed that at 1 week barely 0.01% of cells in the circulation were the transferred cells34. Of the 29 patients that exhibited an objective clinical response, five were complete though only two were ongoing at 21 and 46 months. The median duration of the partial responders was 4 months31.
Emerging information from murine models of ACT emphasized the need for prior lymphodepletion to eliminate regulatory T cells as well as normal endogenous lymphocytes that compete with the transferred cells for homeostatic cytokines23,24,35. Studies using highly selected tumour reactive CD8+ clones administered following lymphodepletion did not result in objective tumour regression, suggesting that the polyclonal nature of tumour reactivity and possibly the presence of CD4+ cells were necessary to mediate tumour rejection3,36. This led to a new generation of ACT clinical protocols with altered techniques for cell growth and with profound host lymphodepletion before cell transfer2,3.
In the latest Surgery Branch, National Cancer Institute trials multiple TIL cultures were initiated from freshly resected metastatic melanomas (a mass of ≥1cm3 was required) and as soon as anti-tumour activity could be detected in vitro against specific unique or shared antigens, cells underwent a rapid expansion using the T-cell-stimulating antibody OKT3 and IL2 (FIG. 1). Approximately 5×1010 cells were infused immediately following a non-myeloablative preparative regimen consisting of 60 mg/kg cyclophosphamide for 2 days followed by 5 days of fludarabine at 25 mg/m2. Twenty-five patients each also received either 2 Gy or 12 Gy plus the chemotherapy regimen. IL2 was administered for 2–3 days at 7.2×105 IU/kg every 8 h.
Figure 1. The generation of anti-tumour T cells used for adoptive cell therapy.
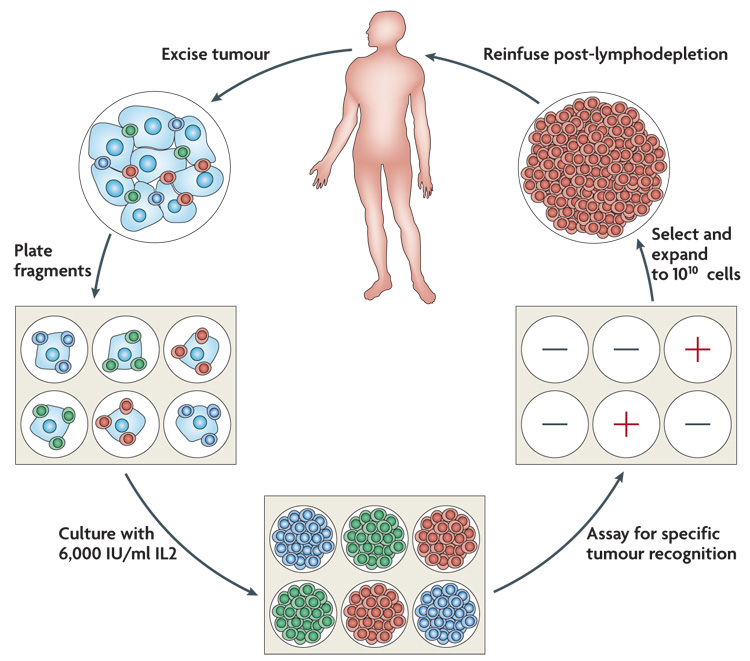
A tumour is excised and multiple individual cultures are established, separately grown and assayed for specific tumour recognition. Cultures with high anti-tumour reactivity are expanded to large numbers (>1010 cells) and reinfused into the cancer patient following the administration of a conditioning lymphodepleting chemotherapy. IL2, interleukin 2.
Objective responses by standard response evaluation criteria in solid tumours (RECIST) were seen in 21 of 43 patients (49%), who received no total body irradiation, in 13 of 25 patients (52%) who received 2 Gy and 18 of 25 patients (72%) who received 12 Gy (TABLE 1). Patients with stable disease were not considered as responders as it is not possible to evaluate this criterion in the absence of randomized patient comparisons. Examples of anti-tumour responses in melanoma patients treated with ACT are shown in FIG. 2. Many of these responses at multiple metastatic sites are durable. None of the 10 complete responders have recurred at times from 8 to 63 months. Fifteen of the partial responders have ongoing responses from 4 to 64 months. The actuarial 3-year survival of patients receiving ACT with the chemotherapy non-myeloablative regimen alone or with 2 Gy total body irradiation is 25% and 42% respectively, compared with 14% for the no lymphodepletion group. The trend for increasing survival as a function of increasing lymphodepletion is highly significant (P = 0.007), but this should be interpreted with caution as this was not a randomized comparison79.
Table 1. Adoptive cell therapy in patients with metastatic melanoma79.
All patients received cyclophosphamide 60 mg/kg for 2 days then fludarabine 25 mg/m2 for 5 days. Includes all patients who received expanded tumour-infiltrating lymphocytes (TIL) plus the full preparative regimen as a first TIL treatment. CR, complete response; OR, objective response; PR, partial response; TBI, total body irradiation.
| Treatment | Patients (n) | Response (n (%)) | ||
|---|---|---|---|---|
| PR | CR | OR | ||
| No TBI | 43 | 17 (39.5) | 4 (9.3) | 21 (48.8) |
| 2 Gy TBI | 25 | 11 (44.0) | 2 (8.0) | 13 (52.0) |
| 12 Gy TBI | 25 | 14 (56.0) | 4 (16.0) | 18 (72) |
Figure 2. Examples of objective tumour regressions in patients receiving adoptive cell transfer of autologous anti-tumour lymphocytes following a lymphodepleting preparative regimen.
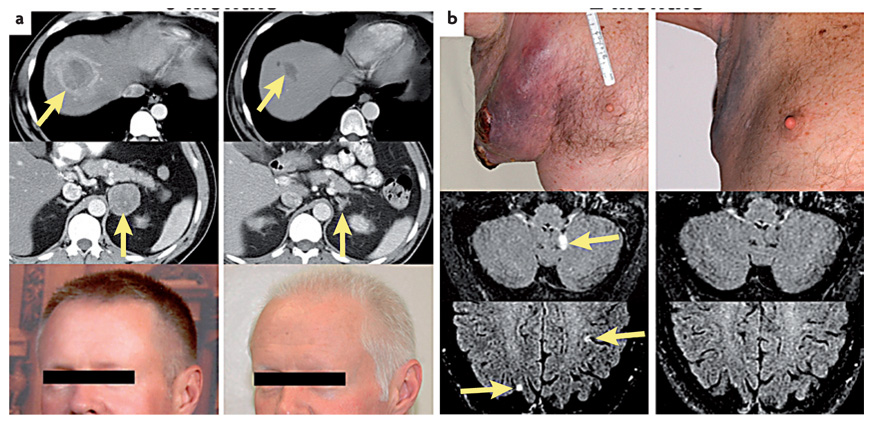
In each case the pretreatment scans and photos are shown on the left and the post-treatment on the right. a | A 45-year-old male with metastatic melanoma to the liver (upper) and right adrenal gland (middle) who was refractory to prior treatment with high dose α interferon as well as high-dose interleukin 2 (IL2). He underwent a rapid regression of metastases and developed vitiligo (lower). b | A 55-year-old male with rapid tumour growth in the axilla as well as multiple brain metastases from metastatic melanoma that was refractory to prior treatment with high dose IL2 who underwent rapid regression of nodal and brain metastases.
There were no added toxicities in these trials owing to the cell administration, although the expected toxicities of IL2 administration were seen most often owing to the capillary leak syndrome caused by IL2.Neutropenic fevers that subsided when white cells recovered were seen in several patients.
Several findings in this trial are of note. The transferred cells expanded in vivo and persisted in the peripheral blood in many patients, sometimes achieving levels of 75% of all CD8+ T cells at 6–12 months after infusion2. Persistence of the transferred T-cell clonotypes correlated with cancer regression37. Eleven of 13 responding patients had ≥5% persistence of transferred clonotypes compared with only 1 of 12 non-responders (P = 0.001). In accord with this, the telomere length of the infused TIL correlated with cancer regression38. The mean telomere length in TIL administered to responding patients was 6.3 kb compared with 4.9 kb in TIL given to non-responders (P < 0.01). TIL clonotypes that persisted in vivo had mean telomeres of 6.2 kb compared with 4.5 kb in non-persisting clonotypes (P < 0.001). Telomere length is related to the proliferative history of the cell. Each time the cell divides telomeres at the ends of chromosomes shorten. Thus cells with longer telomeres have a greater proliferative potential.
In contrast to murine models, the antigen-reactive administered TIL were of effector phenotype, CD27−,CD28−,CD45RA−,CD62L−,CCR7− (REF. 39). By 2 months after transfer, however, circulating tetramer-positive cells had a less differentiated phenotype and were CD27+,CD28+,CDRA+,IL7R− but remained CD62L−,CCR7−, suggesting that highly activated tumour-reactive cells in culture can re-express many of these differentiation markers in vivo39. Re-expression of CD27 in TIL is an indication of a less differentiated cell and, following removal from IL2 in vitro, correlates with the effectiveness of these cells in vivo. These findings are in accord with studies in HIV-positive patients showing that CD27 expression promotes the in vivo survival of administered T cells40.
The high rate of cancer regression seen in these trials convincingly demonstrates the anti-tumour efficacy of ACT therapy and the importance of transferring cells with a high degree of antigen recognition and a high proliferative potential. Other issues of importance that might account for the lack of response in some patients relate to the traffic of the cells to draining lymph nodes or to poor expression of antigens by the tumour. Opportunities for improving ACT for patients with cancer based on the genetic modification of T cells are considered later in this Review.
Other examples of effective ACT therapy
The treatment of patients with cancers expressing viral or alloantigens
When the target antigen on a tumour is ‘foreign’ to the host and the avidity of the T cell is high, as is the case for viral antigens or alloantigens, ACT can be very effective in destroying large tumours in humans. Minor histocompatability antigens such as HA1 and HA2 can represent strong ‘foreign’ targets for effective ACT in cancer patients receiving allogeneic haematopoietic stem cell transplantation (HSC)41. In 1990, Kolb et al. reported cytogenetic remissions in three patients with relapsed chronic myeloid leukaemia treated with buffy coat cells from the HSC marrow donor42. Later studies showed that infusion of donor lymphocytes could mediate complete molecular remissions in 70–80% of patients with relapsed chronic myeloid leukaemia in chronic phase, in 20–30% of patients in blast crisis and in a minority of patients with relapsed multiple myeloma following treatment with allogeneic HSC transplantation43. Cloned CTL lines have induced responses, although the apparent lack of persistence of these lines in vivo has limited their effectiveness44.
The treatment of patients with cancers expressing viral antigens
ACT can mediate cancer regression in humans with Epstein–Barr virus (EBV)-related lymphomas, which occur in patients receiving immunosuppressive drugs. Approximately 90% of humans have lymphocytes latently infected with EBV and the cellular immune response against these viral proteins is essential for the control of infected cells. Approximately 1% of individuals undergoing allogeneic HSC transplants who receive immunosuppressive drugs to prevent graft-versus-host disease develop post-transplant lymphoproliferative diseases (PTLD) in cells of donor origin and as many as 20% of solid organ transplant patients can develop PTLD in cells of recipient origin. These lymphomas express latent EBV antigens, including the immunodominant EBV nuclear antigens (EBNA) EBNA-3A, EBNA-3B and EBNA-3C, that are targets for immunotherapy.
In 1994 O’Reilly and colleagues demonstrated that the infusion of small numbers of normal non-irradiated donor lymphocytes (106/kg) achieved complete responses in five patients with lymphomas occurring following the treatment of leukaemia with chemotherapy and T-cell-depleted allogeneic HSC grafts45. Shortly thereafter, Rooney et al. demonstrated that infusion of long-term cultured EBV-specific T-cell lines generated by repeated in vitro sensitization and expanded in IL2 could effectively prevent and treat PTLD and thus avoid the graft-versus-host disease that is caused by transfer of whole T-cell populations46,47. Donor-derived EBV-specific CTL lines were infused into 60 patients at high risk of developing EBV PTLD and none developed the malignancy, compared with 11.5% of historical controls. Five of six patients treated for overt PTLD experienced a complete regression. These long-term cultured cells could be detected in patients up to 3 years after infusion. These studies have been widely reproduced using EBV-specific donor cell lines to treat donor PTLD in patients undergoing allogeneic HSC transplantation.
PTLD in solid organ transplant patients is a more difficult problem as it is difficult to raise autologous EBV-specific cells in patients on high doses of immuno-suppressive drugs and cells from the organ donor are rarely available48. Thus treatment of these patients has used EBV-specific cell lines from partially matched unrelated (allogeneic) donors49. Recently Haque and colleagues reported a multicentre clinical trial using a bank of 60 long-term allogeneic EBV-reactive T-cell lines to treat PTLD patients. Of 33 patients, 52% achieved a partial or complete response at 6 months after treatment50.
The success in treating EBV PTLD led to the use of autologous anti-EBV cell lines to treat patients with stage 4 nasopharyngeal cancer (NPC) that was refractory to conventional treatments. Virtually all poorly differentiated nasopharyngeal cancer express the less immunogenic EBV latent proteins, LMP-1 and LMP-2, as well as the EBV EBNA-1 antigen. The infusion into six patients of anti-EBV cell lines targeting these antigens led to two complete remissions ongoing at 11 and 23 months and one partial remission lasting 12 months51. In a second series two partial responses were seen in ten treated patients52. These weak EBV antigens are also expressed in the malignant Reed–Sternberg cells found in about 40% of patients with Hodgkin disease. ACT using autologous EBV-reactive cell lines mediated objective responses in 3 of 11 evaluable patients with Hodgkin disease53.
These studies demonstrating the effectiveness of ACT directed against EBV and allogeneic antigens, as well as studies showing that the infusion of IL2-expanded cytomegalovirus (CMV)-reactive autologous T-cell lines grown long-term in IL2 could prevent CMV infection in immunosuppressed patients54, have profound implications for the development of ACT approaches to the treatment of patients with solid cancers. Administration of an avid anti-tumour T cell targeting a highly expressed antigen can result in cancer regression.
ACT using gene-modified lymphocytes
The success of ACT for the treatment of patients with metastatic melanoma has formed a foundation on which to build improvements of this approach. TIL with high avidity for tumour antigens can only be generated from some patients with melanoma and a need exists for the generation of T cells with broad reactivity against shared cancer-associated antigens present on multiple tumour types. The ability to introduce genes into circulating human lymphocytes provides the flexibility to introduce antigen receptors as well as molecules that can provide the cell with enhanced properties required for effective ACT therapy (reviewed in REF 55,REF 56). Genes encoding TCRs can be isolated from high avidity T cells that recognize cancer antigens and retroviral or lentiviral vectors can be used to redirect lymphocyte specificity to these cancer antigens57–60. Mispairing of the inserted chains with endogeneous TCR chains can be greatly reduced by insertion of murine constant region sequences or by insertion of cystine residues that favour pairing of only the transduced chains61,62. High-affinity TCRs can be obtained from rare reactive human clones or from transgenic mouse cells following immunization against human cancer antigens, which thus avoids the tolerance that can limit the generation of these cells in the cancer patient. High-affinity TCR against a p53 epitope63–65 and against carcinoembryonic antigen (M. R. Parkhurst, personal communication) that is present on common epithelial cancers have been generated in transgenic mice and used to redirect the specificity of human lymphocytes. Phage display techniques have been used to generate TCRs with 106× the affinity of a natural TCR directed against the cancer–testis antigen NY-ESO-1, which is expressed on many common cancers66. Chimeric TCR that use the combining site of antibodies genetically fused to intracellular T-cell signalling chains such as CD3ζ can redirect the recognition specificity of lymphocytes to cell surface tumour-associated antigens and thus avoid the limitations of MHC restriction imposed by the use of α–β TCRs67–71.
The steps involved in this process are shown in FIG. 3. High-avidity T cells that are reactive with tumour antigens are identified in the human (often after extensive in vitro sensitization) or from transgenic mice immunized with human cancer antigens. The genes encoding the TCRs from these T cells are cloned and inserted into retroviruses. Retroviral supernatants are then generated under good manufacturing practice conditions that enable their use in humans. These retroviruses can be used to transduce human T cells that express the receptor and can be expanded in vitro for infusion into cancer patients.
Figure 3. The steps involved in generating anti-tumour T cells by inserting genes encoding T-cell receptors.
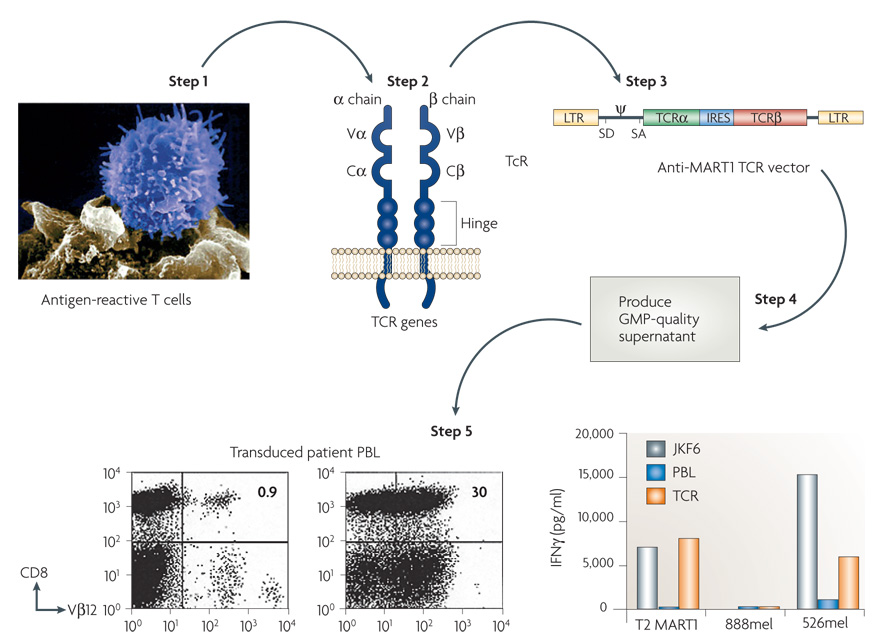
Highly avid anti-tumour T cells are identified and the genes encoding their T-cell receptors (TCRs) are cloned and inserted into retroviruses. Retroviral supernatants are then produced under good manufacturing practice (GMP) conditions and used to insert the T-cell receptors into normal lymphocytes. Expression of the T-cell receptor is then compared in untransduced (UnTd) and transduced (Td) cells by fluorescence-activated cell sorting analysis and by recognition in vitro of HLA-A2+ 526 melanoma line and not the HLA-A2− 888 melanoma line. The effector cells were the anti-MART JKF6 line and untransduced (PBL) and transduced (TCR) lymphocytes. C, constant; IRES, internal ribosome entry site; LTR, long terminal repeat; SA, splice acceptor site; SD, splice donor site; V, variable. Steps 3 and 5 reproduced, with permission, from REF. 4 © American Association for the Advancement of Science (2006).
The first clinical trial to successfully mediate the regression of human cancer by ACT using genetically engineered autologous lymphocytes has recently been published4. Sixteen patients were treated with a TCR that was reactive with the MART1 melanoma antigen isolated from highly reactive TIL. Two patients with metastatic melanoma who received ACT of their autologous normal lymphocytes transduced with genes encoding this MART1 TCR underwent regression of liver and lung hilum metastases respectively and both are currently disease free over 2 years later. Two of 15 additional patients subsequently experienced objective tumour regressions (S.A.R., unpublished observation). TCRs with far greater affinity for the MART1 melanoma antigen have been identified and are now being evaluated in clinical gene therapy trials72. TCRs are now available against a broad array of cancer antigens present on common epithelial cancers and we have recently begun a trial treating patients with epithelial cancers using autologous T cells transduced with a TCR that recognizes a p53 epitope65 (FIG. 4).
Figure 4. Diagram of the retroviral constructs used to insert T-cell receptor (TCR) genes in T cells.
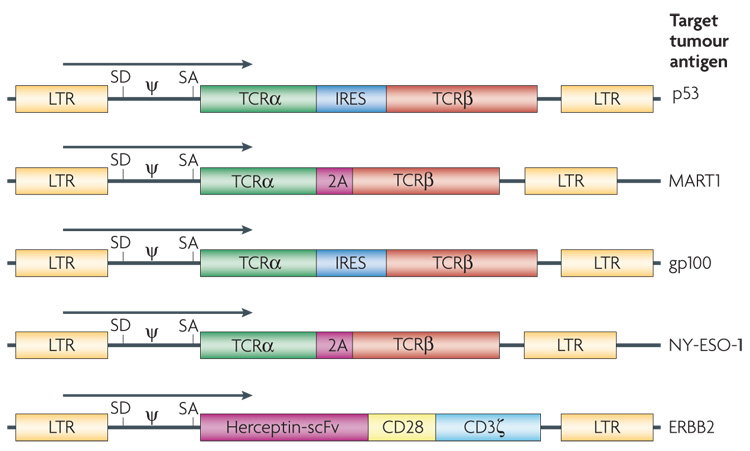
T cells can be engineered with two classes of receptor proteins that are capable of recognizing tumour-associated antigens. Naturally occurring TCRs require coordinated expression of an α and β chain, which can be facilitated by an internal ribosome entry site (IRES) or by the use of a 2A fusion protein. A chimeric antigen receptor is an artificially constructed hybrid protein containing the antigen-binding domains of a single-chain antibody (scFv) linked to T-cell signal domains, such as CD28 and CD3ζ. Vector-specific cis-acting sequences are the long terminal repeat (LTR) that contains the enhancer, promoter and polyadenylation sites, splice donor (SD) and splice acceptor (SA) sequences, and packaging signal (ψ). The target antigen for each of these vectors is as indicated.
As mouse models clearly indicated that increased lymphodepletion could improve the efficacy of ACT12 we are performing clinical trials in which patients receive non-myeloablative chemotherapy plus either 2 Gy or 12 Gy whole body irradiation. Other improvements in ACT are being studied (see TABLE 2 for selected examples). Murine models have shown that CD4+CD25+ FOX3+ regulatory T cells can inhibit ACT and thus the regimen can be modified to deplete CD4+ cells or selectively eliminate T-regulatory cells. Lymphocyte function may be improved using antibodies or genetic approaches that block inhibitory signals on lymphocytes such as CTLA4, PD-1 or TGFβ. Increased persistence and function of the transferred cells may be accomplished by the administration of alternative cytokines such as IL15, or by stimulating the cells in vivo by the administration of a vaccine or by activating host APCs with Toll-like receptor agonists.
Table 2. Opportunities for improving ACT for the treatment of human cancer.
ACT, adoptive cell therapy; APC, antigen-presenting cell; CTLA4, cytotoxic T-lymphocyte-associated 4; IL, interleukin; PD-1, programmed death 1; TCR, T-cell receptor.
| Method | Approach | Example refs |
|---|---|---|
| Genetic modification of lymphocytes to introduce new recognition specificities | αβTCR, chimeric TCR | 4 |
| Genetic modification of lymphocytes to alter function of T cells | Use of co-stimulatory molecules (CD28, 41BB); cytokines (IL2, IL15); homing molecules (CD62L, CCR7); prevention of apoptosis (BCL2) | 76 |
| Modify host lymphodepletion | Selective depletion of CD4+ cells or T regulatory cells | 23 |
| Block inhibitory signals on reactive lymphocytes | Antibodies to CTLA4 or PD-1 | 9 |
| Administer vaccines to stimulate transferred cells | Recombinant virus encoding antigen | 22 |
| Administer alternative cytokines to support cell growth | IL15, IL21 | 77 |
| Stimulate APCs | Use of Toll-like receptor agonists | 27 |
| Generate less differentiated lymphocytes | Alternate culture conditions and growth promoting cytokines in vitro | 26 |
| Overcome antigen escape variants | Use of natural killer cells | 78 |
The future of ACT
In contrast to common epithelial cancers, melanoma appears to be a tumour that naturally gives rise to anti-tumour T cells. However, other cancers are equally susceptible as the targets of reactive T cells. The susceptibility of melanoma to ACT provides optimism for the application of ACT to common epithelial cancers using TCR gene-modified lymphocytes.
A major problem with the application of ACT is that it is a highly personalized treatment and does not easily fit into current modes of oncological practice. The treatment is labour-intensive and requires laboratory expertise. In essence, a new reagent is created for each patient and this patient-specific nature of the treatment makes it difficult to commercialize. Pharmaceutical and biotechnology companies seek off-the-shelf drugs, easy to produce, vial and administer. From a regulatory standpoint, ACT might be more appropriately delivered as a service rather than as a ‘drug’. Blood banks have been instrumental in providing CD34+ haematopoietic stem cells for clinical studies and might be the ideal location for the generation of the anti-tumour T cells needed for ACT.
As modern science increasingly provides the physician with sophisticated information about the unique aspects of an individual cancer, changes in the modes of care delivery need to accommodate this. The ability to use this patient-specific information can lead to a new era of personalized medicine in which individual treatments, such as ACT, are devised for each patient.
Studies of ACT have clearly demonstrated that the administration of highly avid anti-tumour T cells directed against a suitable target can mediate the regression of large, vascularized, metastatic cancers in humans and provide guiding principles as well as encouragement for the further development of immunotherapy for the treatment of patients with cancer.
Glossary
- Adoptive cell therapy (ACT)
The administration of a patient’s own (autologous) or donor (allogeneic) anti-tumour lymphocytes following a lymphodepleting preparative regimen.
- Capillary leak syndrome
The loss of intravascular fluid into soft tissues and lung.
- Objective clinical response
The Response evaluation Criteria in Solid Tumours (RECIST) defines an objective response as a 30% reduction in the sum of the longest diameters of measurable lesions comparing posttreatment with pretreatment values. The World Health Organization criterion defines an objective response to be a 50% reduction in the sum of the products of perpendicular diameters of measurable lesions. In both criteria no new lesions can appear.
- Avidity
The relative intensity of reactivity of lymphocytes when interacting with antigen.
- Allogeneic
Inter-individual genetic variation at the MHC locus. In a partially matched transplant, for example, some MHC antigens are shared by donor and recipient, but in addition the donor has some MHC antigens that the recipient does not.
- Lymphodepletion
Lymphodepletion before ACT uses total body irradiation or cytotoxic drugs to deplete the lymphoid compartment of patients.
- Central memory cells
A subset of antigen-reactive lymphocytes with markers such as CD62L and CCR7 that indicate a less differentiated phenotype.
- Antigen-presenting cells (APC)
A subset of cells that have characteristics enabling them to efficiently present antigenic epitopes to lymphocytes (for example, dendritic cells).
- Non-myeloablative
Relatively modest to moderate doses of chemotherapy are given, not to attack the cancer, but just to suppress the immune system for a brief period of a week or so.
- Effector phenotype
A constellation of cell surface markers that indicate that lymphocytes have differentiated into a mature effector cell capable of recognizing antigen and lysing target cells or secreting cytokines when encountering antigen.
- Alloantigens
An antigen that exists in alternative (allelic) forms in a species, thus inducing an immune response when one form is transferred to members of the species who lack it.
- Buffy coat cells
The plasma layer containing enriched white blood cells that results when whole blood is centrifuged.
- Chronic phase
Indolent phase of the disease in patients with chronic myeloid leukaemia.
- Blast crisis
Aggressive acute phase of the disease in patients with chronic myeloid leukaemia.
- Graft-versus-host disease
Inflammatory and tissue-destructive immune reactions that result from the attack on host tissues by infused allogeneic lymphocytes.
- Post-transplant lymphoproliferative disease (PTLD)
Neoplastic proliferation of lymphocytes that occurs in patients undergoing immunosuppresion, often in preparation for bone marrow or organ transplantation; can occur in host or recipient cells.
- Reed–Sternberg cells
Cells with a characteristic morphology that are thought to be the malignant cells in patients with Hodgkin lymphoma.
- Tolerance
The process that ensures that B- and T-cell repertoires are biased against self-reactivity, reducing the likelihood of autoimmunity.
- Carcinoembryonic antigen
A protein found in fetal gastrointestinal tissue that can be upregulated in some gastrointestinal cancers and can serve as a marker of tumour burden.
- Cancer–testis antigen
A class of antigenic proteins present on some human cancers but not on adult normal tissues except for testes.
Footnotes
DATABASES Entrez Gene: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene
CCR7 | CD25 | CD27 | CD28 | CD34 | CD3ζ | CD4 | CD62L | gp100 | EBNA-1 | EBNA-3A | EBNA-3B and EBNA-3C | FOX3 | IL2 | IL2RA | IL7 | IL7R | IL15 | LMP-1 | MART1 | NY-ESO-1 | p53 | PD-1 | TGFβ
National Cancer Institute: http://www.cancer.gov/
chronic myeloid leukaemia | melanoma | multiple myeloma | nasopharyngeal cancer | renal cancer
National Cancer Institute Drug Dictionary: http://www.cancer.gov/drugdictionary/
References
- 1.Rosenberg SA, et al. Use of tumor infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. Preliminary report. N. Engl. J. Med. 1988;319:1676–1680. doi: 10.1056/NEJM198812223192527.The first paper to demonstrate the regression of cancer using TIL for the immunotherapy of patients with metastatic melanoma.
- 2.Dudley ME, et al. Cancer regression and autoimmunity in patients following clonal repopulation with anti-tumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dudley ME, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J. Clin. Oncol. 2005;23:2346–2357. doi: 10.1200/JCO.2005.00.240.Reference 2 and Reference 3 demonstrate that lymphodepletion prior to ACT can lead to increased cancer regression as well as clonal repopulation of patients with anti-tumour lymphocytes.
- 4.Morgan RA, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003.The first paper demonstrating the adoptive cell transfer of lymphocytes transduced with a retrovirus encoding TCRs that recognize a cancer antigen can mediate anti-tumour responses in patients with metastatic melanoma.
- 5.Rosenberg SA, et al. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N. Engl. J. Med. 1985;313:1485–1492. doi: 10.1056/NEJM198512053132327. [DOI] [PubMed] [Google Scholar]
- 6.Rosenberg SA, Yang JC, White DE, Steinberg SM. Durability of complete responses in patients with metastatic cancer treated with high-dose interleukin-2. Ann. Surg. 1998;228:307–319. doi: 10.1097/00000658-199809000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lotze MT, et al. High dose recombinant interleukin-2 in the treatment of patients with disseminated cancer: responses, treatment related morbidity and histologic findings. J. Am. Med. Assoc. 1986;256:3117–3124. [PubMed] [Google Scholar]
- 8.Kammula US, White DE, Rosenberg SA. Trends in the safety high dose bolus interleukin-2 administration in patients with metastatic cancer. Cancer. 1998;83:797–805. [PubMed] [Google Scholar]
- 9.Phan GQ, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc. Natl Acad. Sci. USA. 2003;100:8372–8377. doi: 10.1073/pnas.1533209100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Attia P, et al. Autoimmunity correlates with tumor regression in patients with metastatic melanoma treated with anti-cytotoxic T-lymphocyte antigen-4. J. Clin. Oncol. 2005;23:6043–6053. doi: 10.1200/JCO.2005.06.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van der Bruggen P, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254:1643–1647. doi: 10.1126/science.1840703. [DOI] [PubMed] [Google Scholar]
- 12.Rosenberg SA. A new era for cancer immunotherapy based on the genes that encode cancer antigens. Immunity. 1999;10:281–287. doi: 10.1016/s1074-7613(00)80028-x. [DOI] [PubMed] [Google Scholar]
- 13.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nature Med. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosenberg SA, et al. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J. Immunol. 2005;175:6169–6176. doi: 10.4049/jimmunol.175.9.6169.The demonstration that peptide vaccines are capable of generating large numbers of anti-tumour lymphocytes in vivo, but these lymphocytes do not appear to have any in vivo ability to prevent recurrence.
- 15.Mitchison NA. Studies on the immunological response to foreign tumor transplants in the mouse. I. The role of lymph node cells in conferring immunity by adoptive transfer. J. Exp. Med. 1955;102:157–177. doi: 10.1084/jem.102.2.157.A seminal paper demonstrating the role of the cellular immune response in the rejection of tumour transplants.
- 16.Delorme EJ, Alexander P. Treatment of primary fibrosarcoma in the rat with immune lymphocytes. Lancet. 1964;2:117–120. doi: 10.1016/s0140-6736(64)90126-6. [DOI] [PubMed] [Google Scholar]
- 17.Fefer A. Immunotherapy and chemotherapy of Moloney sarcoma virus-induced tumors in mice. Cancer Res. 1969;29:2177–2183. [PubMed] [Google Scholar]
- 18.Cheever MA, Kempf RA, Fefer A. Tumor neutralization, immunotherapy, and chemoimmunotherapy of a Friend leukemia with cells secondarily sensitized in vitro. J. Immunol. 1977;119:714–718. [PubMed] [Google Scholar]
- 19.Eberlein TJ, Rosenstein M, Rosenberg SA. Regression of a disseminated syngeneic solid tumor by systemic transfer of lymphoid cells expanded in IL-2. J. Exp. Med. 1982;156:385–397. doi: 10.1084/jem.156.2.385.Demonstration that the intravenous administration of anti-tumour lymphocytes expanded in IL2 could mediate the regression of established disseminated syngeneic tumours in mice.
- 20.Donohue JH, et al. The systemic administration of purified interleukin-2 enhances the ability of sensitized murine lymphocyte to cure a disseminated syngeneic lymphoma. J. Immunol. 1984;132:2123–2128. [PubMed] [Google Scholar]
- 21.Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science. 1986;233:1318–1321. doi: 10.1126/science.3489291.The first demonstration in murine models that the adoptive transfer of TIL could mediate the regression of established murine tumours.
- 22.Overwijk WW, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J. Exp. Med. 2003;198:569–580. doi: 10.1084/jem.20030590.This paper demonstrated that adoptive cell transfer, vaccine and IL2 administration could mediate the rejection of large established transgenic B16 melanomas in mice.
- 23.Antony PA, et al. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J. Immunol. 2005;174:2591–2601. doi: 10.4049/jimmunol.174.5.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gattinoni L, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J. Exp. Med. 2005;202:907–912. doi: 10.1084/jem.20050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wrzesiniski C, et al. Hematopoietic stem cells promote the expansion and function of adoptively transferred antitumor CD8 T cells. J. Clin. Invest. 2007;117:492–501. doi: 10.1172/JCI30414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gattinoni L, et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J. Clin. Invest. 2005;115:1616–1626. doi: 10.1172/JCI24480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paulos CM, et al. Microbial translocation augments the function of adoptively transferred self/tumor-specific CD8+ T cells via TLR4 signaling. J. Clin. Invest. 2007;117:2197–2204. doi: 10.1172/JCI32205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abad JD, et al. T-cell receptor gene therapy of established tumors in a murine melanoma model. J. Immunother. 2008;31:1–6. doi: 10.1097/CJI.0b013e31815c193f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muul LM, Spiess PJ, Director EP, Rosenberg SA. Identification of specific cytolytic immune responses against autologous tumor in humans bearing malignant melanoma. J. Immunol. 1987;138:989–995.The first description of the ability of TIL in the human to recognize human tumour antigens presented on cancer cells.
- 30.Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J. Immunother. 2003;26:332–342. doi: 10.1097/00002371-200307000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosenberg SA, et al. Treatment of patients with metastatic melanoma using autologous tumor-infiltrating lymphocytes and interleukin-2. J. Natl Cancer Inst. 1994;86:1159–1166. doi: 10.1093/jnci/86.15.1159. [DOI] [PubMed] [Google Scholar]
- 32.Aebersold P, et al. Lysis of autologous melanoma cells by tumor infiltrating lymphocytes: association with clinical response. J. Natl Cancer Inst. 1991;13:932–937. doi: 10.1093/jnci/83.13.932. [DOI] [PubMed] [Google Scholar]
- 33.Schwartzentruber DJ, et al. In vitro predictors of therapeutic response in melanoma patients receiving tumor infiltrating lymphocytes and interleukin-2. J. Clin. Oncol. 1994;12:1475–1483. doi: 10.1200/JCO.1994.12.7.1475. [DOI] [PubMed] [Google Scholar]
- 34.Rosenberg SA, et al. Gene transfer into humans: immunotherapy of patients with advanced melanoma, using tumor-infiltrating lymphocytes modified by retroviral gene transduction. N. Engl. J. Med. 1990;323:570–578. doi: 10.1056/NEJM199008303230904. [DOI] [PubMed] [Google Scholar]
- 35.Dummer W, et al. T cell homeostatic proliferation elicits effective antitumor autoimmunity. J. Clin. Invest. 2002;110:185–192. doi: 10.1172/JCI15175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yee C, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc. Natl Acad. Sci. USA. 2002;99:16168–16173. doi: 10.1073/pnas.242600099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robbins PF, et al. Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J. Immunol. 2004;173:7125–7130. doi: 10.4049/jimmunol.173.12.7125.This paper demonstrated that persistence of adoptively transferred cells correlated directly with the likelihood of cancer regression.
- 38.Zhou J, Shen X, Hodes RJ, Rosenberg SA, Robbins P. Telomere length of transferred lymphocytes correlates with in vivo persistence and tumor regression in melanoma patients receiving cell transfer therapy. J. Immunol. 2005;175:7046–7052. doi: 10.4049/jimmunol.175.10.7046.The paper shows that the telomere length of the transferred lymphocytes correlated both with in vivo persistence of the transferred cells as well as with tumour regression.
- 39.Powell DJ, Dudley ME, Robbins PF, Rosenberg SA. Transition of late-stage effector T cells to CD27+ CD28+ tumor-reactive effector memory T cells in humans after adoptive cell transfer therapy. Blood. 2004;101:241–250. doi: 10.1182/blood-2004-06-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ochsenbein AF, et al. CD27 expression promotes long-term survival of functional effector-memory CD8+ cytotoxic T lymphocytes in HIV-infected patients. J. Exp. Med. 2004;200:1407–1417. doi: 10.1084/jem.20040717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marijt WAE, et al. Hematopoiesis-restricted minor histocompatibility antigens HA-1 or HA-2-specific T cells can induce complete remissions of relapsed leukemia. Proc. Natl Acad. Sci. USA. 2003;100:2742–2747. doi: 10.1073/pnas.0530192100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kolb HJ, et al. Donor leukocyte transfusions for treatment of recurrent chronic myelogenous leukemia in marrow transplant patients. Blood. 1990;76:2462–2465.This paper was the first to show that treatment with donor lymphocytes could mediate cytogenetic remissions in patients with chronic myeloid leukaemia.
- 43.Mackinnon S, et al. Adoptive immunotherapy evaluating escalating doses of donor leukocytes for relapse of chronic myeloid leukemia after bone marrow transplantation: separation of graft-versus-leukemia responses from graft-versus-host disease. Blood. 1995;86:1261–1268. [PubMed] [Google Scholar]
- 44.Riddell SR, Bleakley M, Nishida T, Berger C, Warren EH. Adoptive transfer of allogeneic antigen-specific T cells. Biol. Blood Marrow Transplant. 2006;12:9–12. doi: 10.1016/j.bbmt.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 45.Papadopoulos EB, et al. Infusions of donor leukocytes to treat Epstein–Barr virus-associated lymphoproliferative disorders after allogeneic bone marrow transplantation. N. Engl. J. Med. 1994;330:1185–1191. doi: 10.1056/NEJM199404283301703.This paper showed that the infusion of normal donor lymphocytes could achieve complete responses in patients with lymphomas that occurred following the treatment of leukaemia with chemotherapy and T-cell-depleted allogeneic stem cell grafts.
- 46.Rooney CM, et al. Use of gene-modified virus-specific T lymphocytes to control Epstein–Barr-virus-related lymphoproliferation. Lancet. 1995;345:9–13. doi: 10.1016/s0140-6736(95)91150-2. [DOI] [PubMed] [Google Scholar]
- 47.Rooney CM, et al. Infusion of cytotoxic T cells for the prevention and treatment of Epstein–Barr virus-induced lymphoma in allogeneic transplant recipients. Blood. 1998;92:1549–1555.Reference 46 and Reference 47 showed that tumour regression could be obtained by the infusion of long-term cultured EBV-specific T-cell lines.
- 48.Khanna R, et al. Activation and adoptive transfer of Epstein–Barr virus-specific cytotoxic T cells in solid organ transplant patients with posttransplant lymphoproliferative disease. Proc. Natl Acad. Sci. USA. 1999;96:10391–10396. doi: 10.1073/pnas.96.18.10391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Haque T, et al. Complete regression of posttransplant lymphoproliferative disease using partially HLA-matched Epstein–Barr virus-specific cytotoxic T cells. Transplantation. 2001;72:1399–1402. doi: 10.1097/00007890-200110270-00012. [DOI] [PubMed] [Google Scholar]
- 50.Haque T, et al. Allogeneic cytotoxic T cell therapy for EBV-positive post transplant lymphoproliferative disease: results of a phase 2 multicentre clinical trial. Blood. 2007;110:1123–1131. doi: 10.1182/blood-2006-12-063008. [DOI] [PubMed] [Google Scholar]
- 51.Straathof K, et al. Treatment of nasopharyngeal carcinoma with Epstein–Barr virus-specific T lymphocytes. Blood. 2005;105:1898–1904. doi: 10.1182/blood-2004-07-2975. [DOI] [PubMed] [Google Scholar]
- 52.Comoli P, et al. Cell therapy of stage IV nasopharyngeal carcinoma with autologous Epstein–Barr virus-targeted cytotoxic T lymphocytes. J. Clin. Oncol. 2005;23:8942–8949. doi: 10.1200/JCO.2005.02.6195. [DOI] [PubMed] [Google Scholar]
- 53.Bollard C, et al. Cytotoxic T lymphocyte therapy for Epstein–Barr virus Hodgkin’s disease. J. Exp. Med. 2004;200:1623–1633. doi: 10.1084/jem.20040890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Riddell SR, et al. Restoration of viral immunity in immunodeficient humans by the adoptive transfer of T cell clones. Science. 1992;257:238–241. doi: 10.1126/science.1352912. [DOI] [PubMed] [Google Scholar]
- 55.Sadelain M, Riviere I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nature Rev. Cancer. 2003;3:35–45. doi: 10.1038/nrc971. [DOI] [PubMed] [Google Scholar]
- 56.Murphy A, et al. Gene modification strategies to induce tumor immunity. Immunity. 2005;22:403–414. doi: 10.1016/j.immuni.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 57.Cole DJ, et al. Characterization of the functional specificity of a cloned T-cell receptor heterodimer recognizing the MART-1 melanoma antigen. Cancer Res. 1995;55:748–752. [PubMed] [Google Scholar]
- 58.Hughes MS, et al. Transfer of a TCR gene derived from a patient with a marked antitumor response conveys highly active T-cell effector functions. Hum. Gene. Ther. 2005;16:457–472. doi: 10.1089/hum.2005.16.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morgan RA, et al. High efficiency TCR gene transfer into primary human lymphocytes affords avid recognition of melanoma tumor antigen glycoprotein 100 and does not alter the recognition of autologous melanoma antigens. J. Immunol. 2003;171:3287–3295. doi: 10.4049/jimmunol.171.6.3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhao Y, et al. Primary human lymphocytes transduced with NY-ESO-1 antigen-specific TCR genes recognize and kill diverse human tumor cell lines. J. Immunol. 2005;174:4415–4423. doi: 10.4049/jimmunol.174.7.4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res. 2006;66:8878–8886. doi: 10.1158/0008-5472.CAN-06-1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cohen CJ, et al. Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res. 2007;67:3898–3903. doi: 10.1158/0008-5472.CAN-06-3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Theobald MBJ, Dittmer D, Levine AJ, Sherman LA. Targeting p53 as a general tumor antigen. Proc. Natl Acad. Sci. USA. 1995;92:11993–11997. doi: 10.1073/pnas.92.26.11993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kuball J, Schmitz FW, Voss RH. Cooperation of human tumor-reactive CD4+ and CD8+ T cells after redirection of their specificity by a high-affinity p53A2.1-specific TCR. Immunity. 2005;22:117–129. doi: 10.1016/j.immuni.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 65.Cohen CJ, et al. Recognition of fresh human tumor by human peripheral blood lymphocytes transduced with a bicistronic retroviral vector encoding a murine anti-p53 TCR. J. Immunol. 2005;175:5799–5808. doi: 10.4049/jimmunol.175.9.5799.Reference 58–Reference 65 demonstrate that TCRs can be identified that recognize cancer antigens and that transduction of these TCRs into normal human cells can transfer this antigen recognition.
- 66.Li Y, et al. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nature Biotech. 2005;23:349–354. doi: 10.1038/nbt1070. [DOI] [PubMed] [Google Scholar]
- 67.Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl Acad. Sci. USA. 1989;86:10024–10028. doi: 10.1073/pnas.86.24.10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kershaw MH, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 2006;12:6106–6115. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lamers CHJ, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J. Clin. Oncol. 2006;24:e20–e22. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- 70.Park JR, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol. Ther. 2007;15:825–833. doi: 10.1038/sj.mt.6300104. [DOI] [PubMed] [Google Scholar]
- 71.Brentjens RJ, et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nature Med. 2003;9:279–286. doi: 10.1038/nm827. [DOI] [PubMed] [Google Scholar]
- 72.Johnson LA, et al. Gene transfer of tumor-reactive TCR confers both high avidity and tumor reactivity to nonreactive peripheral blood mononuclear cells and tumor-infiltrating. J. Immunol. 2006;177:6548–6559. doi: 10.4049/jimmunol.177.9.6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fernandez-Cruz E, Woda BA, Feldman JD. Elimination of syngeneic sarcomas in rats by a subset of T lymphocytes. J. Exp. Med. 1980;152:823–841. doi: 10.1084/jem.152.4.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Berendt MJ, North RJ. T-cell-mediated suppression of anti-tumor immunity: an explanation for progressive growth of an immunogenic tumor. J. Exp. Med. 1980;151:69–80. doi: 10.1084/jem.151.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mule JJ, Shu S, Schwarz SL, Rosenberg SA. Adoptive immunotherapy of established pulmonary metastases with LAK cells and recombinant interleukin-2. Science. 1984;225:1487–1489. doi: 10.1126/science.6332379. [DOI] [PubMed] [Google Scholar]
- 76.Liu K, Rosenberg SA. Transduction of an interleukin-2 gene into human melanoma-reactive lymphocytes results in their continued growth in the absence of exogenous IL-2 and maintenance of specific antitumor activity. J. Immunol. 2001;167:6356–6365. doi: 10.4049/jimmunol.167.11.6356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Waldmann TA. The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nature Rev. Immunol. 2007;6:595–601. doi: 10.1038/nri1901. [DOI] [PubMed] [Google Scholar]
- 78.Ruggeri L, Mancusi A, Capanni M, Martelli MF, Velardi A. Exploitation of alloreactive NK cells in adoptive immunotherapy of cancer. Curr. Opin. Immunol. 2005;17:211–217. doi: 10.1016/j.coi.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 79.Dudley ME, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J. Clin. Oncol. doi: 10.1200/JCO.2008.16.5449. (in the press). [DOI] [PMC free article] [PubMed] [Google Scholar]