

Abstract
Rationale
Cardiac fibrosis contributes to pathogenesis of atrial fibrillation (AF), which is the most sustained arrhythmia and a major cause of morbidity and mortality. Although it has been suggested that Ca2+ signals are involved in fibrosis promotion, the molecular basis of Ca2+ signaling mechanisms and how Ca2+ signals contribute to fibrogenesis remain unknown.
Objective
To determine the molecular mechanisms of Ca2+-permeable channel(s) in human atrial fibroblasts, and to investigate how Ca2+ signals contribute to fibrogenesis in human AF.
Methods and Results
We demonstrate that the transient receptor potential melastatin related 7 (TRPM7) is the molecular basis of the major Ca2+-permeable channel in human atrial fibroblasts. Endogenous TRPM7 currents in atrial fibroblasts resemble the biophysical and pharmacological properties of heterologous expressed TRPM7. Knocking down TRPM7 by small hairpin RNA (shRNA) largely eliminates TRPM7 current and Ca2+ influx in atrial fibroblasts. More importantly, atrial fibroblasts from AF patients show a striking upregulation of both TRPM7 currents and Ca2+ influx and are more prone to myofibroblast differentiation, presumably due to the enhanced expression of TRPM7. TRPM7-shRNA markedly reduced basal AF fibroblast differentiation. Transforming growth factor β1 (TGF-β1), the major stimulator of atrial fibrosis, requires TRPM7-mediated Ca2+ signal for its effect on fibroblast proliferation and differentiation. Furthermore, TGF-β1 induced differentiation of cultured human atrial fibroblasts is well correlated with an increase of TRPM7 expression induced by TGF-β1.
Conclusions
Our results establish that TRPM7 is the major Ca2+-permeable channel in human atrial fibroblasts, and likely plays an essential role in TGF-β1-elicited fibrogenesis in human AF.
Keywords: Atrial Fibrillation, TRPM7, Ca2+ signaling, TGF-β1, fibrogenesis
INTRODUCTION
Cardiac fibrosis is a detrimental factor that results in abnormalities in cardiac conduction, stiffening of the ventricular walls, and loss of contractility, thereby contributing to a variety of heart diseases, including hypertrophy, heart failure, and arrhythmia 1. Atrial fibrillation (AF) is the most sustained clinical arrhythmia and is a major cause of morbidity and mortality 2. Recent studies have demonstrated that structural remodeling as a result of fibrosis is one of the fundamental mechanisms underlying the perpetuation of AF, and contributes synergistically with electrical and contractile remodeling to AF substrate 2-6. Atrial fibrosis is a hallmark feature of arrhythmogenic structural remodeling in clinical AF 7. Increased amount of fibrosis occurs in not only AF patients associated with other cardiac diseases 8, 9, but also in those with lone AF 8, 10. There is a positive correlation between the amounts of fibrosis and the persistence of AF 9, suggesting that atrial fibrosis results in structural remodeling to promote AF. The causal importance of fibrosis in occurrence and persistence of AF is strongly supported by studies in animal models in which selective atrial fibrosis caused by overexpression of TGF-β1 increases AF vulnerability 11, 12. Thus, understanding the mechanism of fibrogenesis is important in developing new therapeutic strategies aimed at preventing or reversing structural remodeling in AF.
Fibrosis represents excessive deposition of extracellular matrix proteins (ECM) synthesized by cardiac fibroblasts or myofibroblasts upon stimulation 13, 14. Myofibroblasts are not found in normal cardiac tissue 15. In response to pathological stimuli, such as myocardial injury, oxidative stress, mechanical stretch, autocrine-paracrine mediators, and inflammatory stimuli, fibroblasts proliferate, migrate and undergo phenotypic changes and differentiate into myofibroblasts 14, 16. Myofibroblasts might also be derived from endothelial cells in fibrotic heart 17. By producing growth factors, cytokines, chemokines, ECM proteins, and proteases 18, 19, myofibroblasts play a pivotal role in fibrosis. Inhibition of myofibroblast differentiation may prove to be a common and effective approach to attenuate cardiac fibrosis, regardless of the initial stimuli. However, little is known about the molecular mechanisms underlying fibroblast differentiation.
Ca2+ signals are essential for diverse cellular functions including differentiation, gene expression, cell proliferation, growth and death 20. In cardiac fibroblasts, several lines of evidence suggest that Ca2+ entry is essential for fibroblast functions. Chelating external Ca2+ by EGTA prevents substance P induced proliferation of cultured rat cardiac fibroblasts 21, 22. Eliminating external Ca2+ to prevent Ca2+ influx attenuates H2O2 induced IL-6 mRNA expression 23. In mechanical-stimulation or stretch-induced membrane potential studies, both Ca2+-entry and Ca2+-release are involved in generation of MIP (MIP: mechanically induced potential) 24, 25. A stretch induced current can be blocked by Gadlinium (Gd3+) 26, a non-selective cation channel blocker. Recently, it has been shown that intracellular Ca2+ contributes to Angiotensin II (Ang II) induced proliferation of cardiac fibroblasts 27. In an in vivo study, Mibefradil, a Ca2+ channel blocker, significantly reduced collagen production and fibroblast differentiation in rats treated with AngII or Aldosterone 28. These studies indicate that Ca2+ entry through Ca2+-permeable ion channels is essential for gene expression and fibrosis promotion. Therefore, understanding the molecular basis of Ca2+-permeable channels is crucial for elucidating the molecular mechanisms of proliferation and differentiation of cardiac fibroblasts.
Fibroblasts have been reported to have depolarized resting membrane potentials. Measured by standard microelectrode techniques in multicellular tissues, the resting membrane potential of atrial fibroblasts is between -31 to -16 mV 29-31. There is no evidence indicating that functional voltage-gated calcium channels exist in the fibroblast 32. The transient receptor potential (TRP) channels are responsible for Ca2+ entry in various non-excitable and excitable cells 33-35. TRP channels are non-voltage gated but are activated by a variety of different stimuli including receptor activation, oxidative stress, mechanical stretch, cell metabolites, and thermal or sensory stimuli 33-35. TRP channels also enable individual cells to sense changes in their local environment. Given that cardiac fibroblasts encounter a variety of pathological conditions, it seems that TRP channels could be the potential candidates for Ca2+ signaling in cardiac fibroblasts.
In order to understand Ca2+ signaling mechanisms in cardiac fibroblasts and potential roles of Ca2+ signals in the cardiac fibrogenesis cascade, we investigated the molecular basis of Ca2+-permeable channels in human atrial fibroblasts isolated from AF patients and normal sinus rhythm patients (NSR), and studied how Ca2+ influx contributes to TGF-β1 induced fibrogenesis process. We discovered that TRPM7, a Ca2+-permeable cation channel which also possesses protein kinase function 36-38, underlies the Ca2+ signaling mechanism in human atrial fibroblasts. TRPM7 has been reported to play a vital role in embryonic development 39 and anoxic cell death 40. We found that TRPM7 is the major Ca2+-permeable channel in human atrial fibroblasts. Knocking down TRPM7 by shRNA largely eliminates the endogenous TRPM7 currents as well as Ca2+ influx in atrial fibroblasts. Strikingly, TRPM7 and TRPM7-medaited Ca2+ influx are drastically up-regulated in fibroblasts from AF patients. In addition, inhibition of TRPM7-mediated Ca2+ influx renders fibroblasts less sensitive to TGF-β1 induced proliferation and differentiation, indicating that TRPM7-mediated Ca2+ signal is necessary for TGF-β1 elicited fibrogenesis. On parallel with myofibroblast differentiation, TRPM7 is up-regulated by TGF-β1 in cultured fibroblasts. Taken together, our results indicate that TRPM7-mediated Ca2+ signal contributes to fibroblast differentiation and may mediate the fibrogenic effect of TGF-β1. This study suggests that inhibition of TRPM7 may prove to be an effective approach to reduce fibroblast differentiation and therefore attenuate fibrosis during human AF.
METHODS
Human Cardiac Tissue Sample Collection and fibroblasts isolation
Myocardial samples of the right atria were collected during cardiac surgery. All procedures involving human tissue use were approved by the institutional review boards of the University of Connecticut Health Center. Atrial fibroblasts were isolated by collagenase (see details in the supplementary methods). Freshly isolated fibroblasts from AF and NSR patients were used for patch-clamp experiment to compare the TRPM7 current amplitude.
Molecular Biology
RT-PCR was performed as we previously reported 41 by using the primers shown in Online Table II. TRPM7-specific shRNAs were designed and constructed into lenti-viral delivery system. The efficiency of the shRNAs was tested in the TRPM7 over-expression HEK-293 cells. The one with higher efficiency was used in the experiments (see the Online Data Supplement)
Electrophysiology
Whole-cell and single-channel currents were recorded as we reported previously 42, 43. Internal Mg2+-free solution was used for whole-cell current recording and inside-out configuration was applied for single channel current recording. For details, see the Online Data Supplement.
Ratio Ca2+ imaging experiments
Ca2+ influx was measured using ratio Ca2+ imaging system and analyzed using NIS-Elements (Nikon). The F340/F380 ratio in the presence of 20 mM Ca2+ was normalized to that of 1 μM Ionomycin (Iono) elicited Ca2+ signal (see details in supplementary methods). Ca2+ influx was also measured with/without store depletion by 2 μM thapsigargin (Tg) in some experiments as specified in text.
Immunostaining
Fibroblasts were fixed by 4% formaldehyde and immmunostained with α-SMA antibody. The immunostained cells were analyzed using a Zeiss LSM 510 confocal microscope. Details are described in the Online Data Supplement.
Western Blot
The protein expression levels of TRPM7 and α-SMA were evaluated by Western blot experiments. GAPDH was used as a loading control (the Online Data Supplement).
Data Analysis
Pooled data are presented as mean ± SEM. Dose–response curves were fitted by an equation of the form E=Emax{1/[1+(EC50/C)n]}, where E is the effect at concentration C, Emax is maximal effect, EC50 is the concentration for half-maximal effect and n is the Hill coefficient. Statistical comparisons were made using two-way ANALYSIS of variance (ANOVA) and two-tailed t-test with Bonferroni correction. Fisher’s exact test was used for analysis of percentage differences for patients’ database. P < 0.05 indicated statistical significance.
RESULTS
TRPM7 is the major Ca2+-permeable channel in human atrial fibroblasts
To understand which Ca2+ permeable channel is responsible for Ca2+ entry in cardiac fibroblasts, we first investigated the expression of Ca2+-permeable TRP channels. Among the 27 human TRP channel genes 33, we found that TRPM7 is abundantly expressed in human atrial fibroblasts (Online Figure I). To study whether TRPM7 constitutes functional channels in the fibroblasts, we applied voltage-clamp to investigate the current characteristics. As shown in Figure 1, a TRPM7-like current was readily elicited by a voltage ramp protocol ranging from -120 to +100 mV. The current-voltage relation (I-V relation) with small inward current and strong outward rectification (Figure 1A) is similar to the typical characteristics of the recombinant TRPM7 36, 37. Heterologous expressed TRPM7 exhibits several unique features, including single channel conductance, potentiation by low external pH, inhibition by low concentrations of 2-aminoethoxydiphenyl borate (2-APB) but potentiation by high concentration of 2-APB 43, 44. We then tested if the endogenous TRPM7-like current in cardiac fibroblasts has similar properties. The single channel conductance of TRPM7-like current (Figure 1B) is similar to that of TRPM7 over-expressed in CHO cells 43. TRPM7-like current was significantly potentiated by low extracellular pH (Figure 1C) with an EC50 of 3.9 pH unit (Online Figure IIB). Like TRPM7, TRPM7-like current was inhibited by low concentrations of 2-APB with an IC50 of 32.1 μM (Figure 1E-F). High concentrations of 2-APB enhanced TRPM7-like current (Figure 1E), but the concentration-dependence of the effect was not assessed because of poor solubility of 2-APB at high concentrations. All of these features of the endogenous TRPM7-like currents in human atrial fibroblasts are indistinguishable from those of exogenously expressed TRPM7 42, 43, suggesting that the TRPM7-like current in the human cardiac fibroblasts is encoded by TRPM7.
Figure 1.

The endogenous TRPM7-like channels in human atrial fibroblasts resemble the characteristics of recombinant TRPM7. A, Representative TRPM7-like currents elicited by a voltage ramp protocol ranging from -120 to +100 mV. B, Single channel conductance of TRPM7-like channel was 38.4 ± 1.6 pS (n=6). Representative recordings from inside-out patches are shown in the inset. C, TRPM7-like inward current was potentiated by external acidic pH solution. D, Concentration-dependent effects of external protons on TRPM7-like currents. E, Biphasic effects of 2-APB on TRPM7-like currents, with inhibition and potentiation at low and high 2-APB concentrations, respectively. F, Concentration-dependent inhibition of 2-APB on TRPM7-like currents. Best fit with Hill equation yielded IC50=32.1 ± 0.1 μM, nH=1.1 (mean ± SEM, n=5).
To further confirm that TRPM7 constitutes TRPM7-like currents, we applied TRPM7-specific shRNA to the fibroblasts (Figure 2). The efficiency of TRPM7-shRNA was first examined in HEK-293 cells over-expressing human TRPM7 (Figure 2A-B). The current amplitude of TRPM7 was decreased by ~70% after TRPM7-shRNA treatment. Similarly, after treatment for 5 days, the current amplitude of TRPM7-like currents in human fibroblasts was significantly reduced by TRPM7-shRNA in comparison with the cells treated with Scr-shRNA (Figure 2C-D), indicating that TRPM7 is indeed the molecule encoding TRPM7-like currents in human atrial fibroblasts. Based on these findings, we refer to the TRPM7-like currents as “TRPM7” in the following text.
Figure 2.

TRPM7 underlies the molecular basis of the TRPM7-like channel and that TRPM7-like channel mediated Ca2+ entry constitutes the major Ca2+ influx in human atrial fibroblasts. A, Typical recordings of TRPM7 over-expressed in HEK-293 cells after treatment with scramble (Scr)-shRNA and TRPM7-shRNA. B, Average current amplitude of TRPM7. TRPM7-shRNA decreased TRPM7 by about 70%. C, Current amplitude of TRPM7-like current recorded in human atrial fibroblasts in a TRPM7-specific shRNA treated cell (Blue) in comparison with that of a Scr-shRNA treated cell (Red). D, Mean current density of TRPM7-like current in TRPM7-shRNA and Scra-shRNA treated cells. E, Ca2+ influx mediated by TRPM7-like channel. Ca2+ influx was elicited by 20 mM Ca2+. The amplitude of Ca2+ influx was normalized to the peak Ca2+ influx induced by 1 μM Ionomycin (Iono). TRPM7-shRNA significantly reduced the Ca2+ influx (Blue) in comparison with the Ca2+ influx obtained in scramble shRNA treated cells (Red). F, TRPM7 shRNA reduced Ca2+ influx in human atrial fibroblasts by 62% measured by normalized peak amplitude.
TRPM7 is responsible for Ca2+entry in human atrial fibroblasts
We next asked whether TRPM7 is responsible for Ca2+ entry in atrial fibroblasts. Using ratio Ca2+ imaging measurements, we found that Ca2+ influx was decreased by 62% after fibroblasts were treated with TRPM7-shRNA for 5 days, indicating that TRPM7 is the major Ca2+-permeable channel in human atrial fibroblasts (Figure 2E-F). To elucidate whether there are other Ca2+-permeable channels which may contribute to Ca2+ influx in human atrial fibroblasts, we used the conditions specific for TRPV2 45, TRPV4 46 or TRPC6 47, 48 current recording, and tested if functional currents of these channels are present in human atrial fibroblasts. As shown in Figure 3, TRPV2-like currents could not be elicited by 200 μM 2-APB in human atrial fibroblasts (Figure 3A), whereas 2-APB readily activated TRPV2 overexpressed in HEK293 cells (Figure 3B-C). The conditions used for activating TRPV4 expressed in HEK293 cells (Figure 3E-F) failed to induce any TRPV4-like current in atrial fibroblasts. TRPC6 can be elicited by activation of Gq-like receptor activation (Figure 3H-I). However, neither using LPA to activate EDG7 receptor (Figure 3G) nor including GTPγS in the pipette solution (Online Figure III) was effective in inducing TRPC6-like current in atrial fibroblasts. These results, summarized in Figure 3, indicate that TRPV2, TRPV4, and TRPC6 do not constitute functional currents even though their expression was detectable by RT-PCR (Online Figure I). Therefore, it is unlikely that TRPV2, TRPV4 or TRPC6 contribute to Ca2+ entry in human atrial fibroblasts.
Figure 3.

TRPV2, TRPV4 and TRPC6 currents were not detectable in atrial fibroblasts. A, TRPV2-like current was not elicited by 2-APB (200 μM) in human atrial fibroblasts. B, Typical recording of TRPV2 over-expressed in HEK293 cells. C, Time-dependent changes of inward and outward currents of TRPV2 in HEK293 cells. D, 4α-PDD (1 μM) failed to induce TRPV4-like current in human atrial fibroblasts. E, Representative recording of recombinant TRPV4 activated by 1 μM 4α-PDD in HEK293 cells. F, Time-dependent changes of inward and outward currents of TRPV4 in HEK293 cells. G, Activation of Gq-linked EDG7 receptors in fibroblasts by Oleoyl-lysophosphatidic acid (LPA) could not activate TRPC6-like current. H, Recombinant TRPC6 current activated by Carbachol (CCh) in HM1 cells (HEK293 cells with over-expression of type 1 muscarinic receptor). I, Time-dependent changes of inward and outward TRPC6 currents in HM1 cells. Please note that the currents recorded in the beginning (in A, D, and G) were endogenous TRPM7 which were inhibited by Mg2+ when the pipette solution dialyzed into the cell. Inward and outward currents in C, F and I were measured at -100 and +100 mV respectively.
It appears that TRPC1 is abundantly expressed at mRNA level (Online Figure I) in human atrial fibroblasts. Since TRPC1 may form heteromeric channels with TRPC4 or TRPC5 49, we attempted to record heteromeric currents with I-V relations similar to those of TRPC1/TRPC4 (or TRPC1/TRPC5). However, including GTPγs in the pipette solution failed to induce any TRPC1/TRPC4- or TRPC1/TRPC5-like current (data not shown), suggesting that TRPC1 may not be able to form functional heteromeric channels in atrial fibroblasts. Knocking down hTRPC1 by TRPC1 siRNA did not influence Ca2+ influx or fibroblast differentiation (Online Figure IV), further indicating that TRPC1 may not be able to contribute Ca2+ influx in human atrial fibroblasts.
We next investigated whether store depletion-activated Ca2+ channel (ICRAC) may influence Ca2+ entry in atrial fibroblasts. As shown in Figure 5, store-depletion by 5 μM thapsigargin (Tg) did not induce any current, suggesting that store depletion activated channels are not functionally expressed in human atrial fibroblasts. Moreover, Ca2+ influx was not changed by Tg, further suggesting that store-depletion does not contribute to Ca2+ signals in human atrial fibroblasts. We also investigated whether TRPV2, TRPV4, TRPC1/TRPC4, TRPC6 and ICRAC currents are present in fibroblasts from AF patients, and found that TRPM7 current is the only current that we were able to record in fibroblasts from both NSR and AF patients. Therefore, it appears that endogenous TRPM7 is the major channel which mediates Ca2+ entry in human atrial fibroblasts.
Figure 5.

Increased Ca2+ influx in AF fibroblasts in comparison with that in NSR fibroblasts. A, Changes of F340/F380 ratio under indicated conditions. There was more Ca2+ influx in the fibroblasts from AF patients when the cells were perfused with 20 mM Ca2+. Ca2+ influx was measured after the fibroblasts were cultured for 12 hrs which allows fibroblasts to attach to the coverslips. B, Normalized F340/F380 ratio from a representative experiment using the cells from AF and NSR patients. C, Average ratio of F340/F380 in fibroblasts from AF and NSR patients. The ratio was normalized to the maximal response induced by 1 μM ionomycin.
TRPM7 is markedly up-regulated in atrial fibroblasts from AF patients
It has been shown that fibrosis associated structural remodeling is a major contributor to AF 7. However, the Ca2+ signaling mechanism in fibrogenesis is unknown. Our findings that TRPM7 is the major Ca2+-permeable channel in human atrial fibroblasts provided us an excellent opportunity to explore how Ca2+ signals are involved in fibrosis associated AF. The left atrial size of AF patients was similar to that in NSR patients among those enrolled in the current study (Online Table I), although a larger group of AF and NSR patients, including those who were not enrolled in the fibroblast study, showed a larger left atrial size in the AF vs. NSR patients (data not shown). We used freshly isolated fibroblasts from biopsy specimens for current recording. This eliminated cell culture related factors which could interfere with the experimental results. We found that TRPM7 currents recorded from freshly isolated fibroblasts in AF patients were 3- to 5-fold larger than those from NSR patients (Figure 4). Both inward and outward currents were increased significantly. Since measurement of outward current amplitude gives a more accurate assessment, we used outward current amplitude measured at +100 mV for comparison (Figure 4C-D). The average TRPM7 current density was 32.5 ± 2.3 pA/pF in fibroblasts from AF patients (88 cells from 7 patients) and 11.1±0.5 pA/pF in fibroblasts from NSR patients (87 cells from 8 patients), respectively. The increase in TRPM7 current density appears to be resulted from increased membrane expression of TRPM7 channel proteins, as the single channel conductance and open probability of TRPM7 were indistinguishable between the NSR fibroblasts and AF fibroblasts (Online Figure VI). We were not able to do western blot experiments to determine TRPM7 protein levels in NSR and AF fibroblasts due to the limited amount of tissues and limited number of fibroblasts. However, we observed that whereas we were able to obtained single channel recordings from more than 95% of patches in the AF fibroblasts, less than 50% of patches from NSR fibroblasts displayed single channel openings, suggesting that membrane protein level of TRPM7 was higher in AF fibroblasts than that in NSR fibroblasts.
Figure 4.

Up-regulation of endogenous TRPM7 in atrial fibroblasts from AF patients. A, Typical TRPM7 recording in a freshly isolated fibroblast from AF patient. B, TRPM7 recorded in a freshly isolated fibroblast from NSR patient. C, Average current density of TRPM7 from each AF and NSR patient. Note that current amplitudes from all the AF patients are larger than the biggest current amplitude in NSR patients. D, Average current density of endogenous TRPM7 from all the fibroblasts in AF and NSR patients.
To study if the increased TRPM7 currents result in enhanced Ca2+ influx, we used ratio Ca2+ imaging method to compare the Ca2+ entry through TRPM7 in fibroblasts isolated from AF and NSR patients. Since TRPM7 channels conduct small inward currents and are constitutively active, we used 20 mM Ca2+ in order to obtain better signal/noise ratio. In addition, cells were briefly exposed to a nominally divalent free solution (DVF) in order to eliminate any blockade of TRPM7 by external Mg2+. As shown in Figure 5, the peak Ca2+ influx in fibroblasts from AF patients was 10-fold greater than Ca2+ influx in the fibroblasts from NSR patients. This result not only further indicates that TRPM7 is the major Ca2+-permeable channel responsible for Ca2+ influx in human atrial fibroblasts, but also strongly implies that TRPM7-mediated Ca2+ signals may play an important role in AF-associated fibrogenesis.
TRPM7 is required for differentiation of human atrial fibroblasts
As the myofibroflast is the major cell type that synthesizes growth factors, cytokines, chemokines, and extracellular matrix (ECM) proteins, and plays a pivotal role in fibrogenesis 13, 19, we tested whether there was a higher percentage of myofibroblasts in the cells isolated from atrial samples from AF patients than those from NSR patients. As shown in Figure 6, after 24 hrs culture, without TGF-β1 treatment, cells from AF patients displayed 44% myofibroblasts, whereas only 16% cells from NSR patients were myofibroblats. Such data suggest that fibroblasts from AF patients are more prone to differentiation into myofibroblasts. The increased vulnerability to differentiation in the fibroblasts from AF patients might result from an up-regulated TRPM7 level. After TGF-β1 (10 ng/ml) induction, 90% of fibroblasts differentiated into myofibroblasts in both NSR and AF groups.
Figure 6.

Myofibroblast differentiation in cells from AF and NSR patients. A, Immunostaining with antibody against α-SMA in fibroblasts from NSR and AF patients, respectively. Fibroblasts from AF patients show a higher percentage of differentiated myofibroblasts in the absence of TGF-β1treatment (P <0.01). TGF-β1(10 ng/ml) treatment induced 90% differentiated myofibroblasts in cells from both AF and NSR patients. α-SMA is in red, and nuclei are labeled in blue by TO-PROIII. B, Mean percentage of differentiated myofibroblasts in AF or NSR patients with or without TGF-β1treatment. The basal differentiation (without TGF-β1treatment) in AF patients is higher than that in NSR patients (P <0.01). C, TRPM7-shRNA decreased Ca2+ inlux in atrial fibroblasts from AF patient . The average decrease of Ca2+ influx by TRPM7-shRNA was 64%. D, The basal differentiation of atrial fibroblasts from AF patient was significantly decreased by TRPM7-shRNA.
To determine the contribution of TRPM7 to basal differentiation of fibroblasts from AF patients, AF fibroblasts were treated with TRPM7-shRNA or Scr-shRNA in 1% FBS media after atrial fibroblasts attached to the coverslips (by culturing for 12 hrs in 10% FBS media) for 72 hrs. Fibroblasts were then cultured in 10% FBS media for 24 hr, followed by Ca2+ influx and differentiation assessment. As shown in Figure 6C-D, TRPM7-shRNA significantly decreased Ca2+ influx and inhibited basal AF fibroblast differentiation, indicating that TRPM7-mediated Ca2+ entry contributes to basal differentiation of AF fibroblasts.
We further investigated whether TRPM7 is involved in TGF-β1 induced differentiation. Human atrial fibroblasts from NSR patients were treated with TRPM7-shRNA and scramble shRNA (Scr-shRNA). TGF-β1 was then applied to induce differentiation after cells were serum starved for 24 hours. As shown in Figure 7, without TGF-β1 (10 ng/ml) treatment, there were 30.0% myofibroblasts in the Scr-shRNA treated group and 25.4% myofibroblasts in the TRPM7-shRNA treated group. This higher basal differentiation of NSR fibroblasts in comparison with that in Fig. 6 was due to that the fibroblasts were culture in 10% FBS media for more than 84 hrs, and there is a time-dependent increase in basal differentiation rate when fibroblasts were cultured in 10% FBS media (Online Figure VII). After TGF-β1 stimulation, the differentiated myofibroblasts in the Scr-shRNA treated group increased to 61.0%, whereas, knocking down of TRPM7 by TRPM7-shRNA significantly reduced the differentiated myofibroblasts to 34.4%, a level similar to the control group (Figure 7B). The loss of sensitivity to TGF-β1 in TRPM7-shRNA treated fibroblasts suggests that TRPM7 plays a role in TGF-β1 induce atrial fibroblast differentiation.
Figure 7.
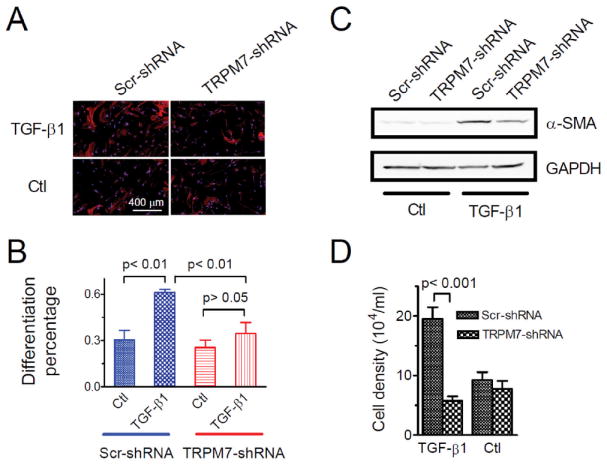
Effects of TRPM7 on fibroblast differentiation and proliferation. A, Differentiated myofibroblasts detected by immunostaining of α-SMA (Red). Human atrial fibroblasts were isolated from NSR patients and treated with Scr- or TRPM7-shRNA. Differentiation was induced by 10 ng/ml TGF-β1. Differentiation percentage was obtained by counting the number of differentiated cells (Red) divided by the total number of cells as identified by the nuclei (Blue). B, Average percentage of differentiated myofibroblasts in Scr- or TRPM7-shRNA treated cells. In TRPM7-shRNA treated cells, differentiation induced by TGF-β1was not significantly different from that of cells without TGF-β1 induction (34.4 ± 7.2% and 23.5± 4.8% respectively; p >0.05). In Scr-shRNA treated cells, the differentiation induced by TGF-β1 (61.0 ± 1.9%) was significantly higher than that in control cells (30.4 ± 6.1%). C, Changes in α-SMA induced by 10 ng/ml TGF-β1 in TRPM7- or Scr-shRNA treated mouse cardiac fibroblasts. TGFβ receptor blocker SB431542 (10 μM) was applied to the control groups to minimize differentiation at basal level. α-SMA protein identified by α-SMA antibody; GAPDH was used as loading control. Please note the significant decrease of α-SMA in TRPM7-shRNA treated cells. Similar results were obtained in three separate experiments. D, Effects of TRPM7-shRNA on human atrial fibroblast proliferation induced by TGF-β1 (10 ng/ml).
To understand how TRPM7 may be involved in fibroblast differentiation, we quantified changes of α-SMA protein in scramble- and TRPM7-shRNA treated cells by Western blot. This experiment was performed by using mouse fibroblasts due to the limited number of human fibroblasts. To rule out other factors such as residual TGFβ 50 in serum which may influence differentiation, we applied the TGF-β1 receptor blocker SB431542 as a control for TGF-β1 treatment. As shown in Figure 7C, the increased protein expression of α-SMA induced by TGF-β1 was significantly reduced by TRPM7 shRNA, which is consistent with the immunostaining result in human fibroblasts (Figure 7A-B).
Role of TRPM7 in TGF-β1 induced fibroblast proliferation
We next investigated the role of TRPM7 in proliferation of fibroblasts. Human cardiac fibroblasts treated with scramble or TRPM7-shRNA were exposed to TGF-β1 (10 ng/ml). As shown in Figure 7D, TRPM7-shRNA significantly decreased the number of fibroblasts, suggesting that TRPM7 is also involved in cardiac fibroblast proliferation caused by TGF-β1.
Regulation of TRPM7 by TGF-β1
As TGF-β1 is the most potent fibrogenesis stimulator, we studied whether expression of TRPM7 is modulated by TGF-β1. In this experiment, fibroblasts were treated with TGF-β1 or the TGF-β1 receptor blocker SB431542. TRPM7 currents were recorded after the human atrial fibroblasts were treated for 24 hr. As shown in Figure 8A-B, TRPM7 current density was increased by about 80% by TGF-β1. Ca2+-influx in TGF-β1 treated fibroblasts was significantly larger than that of control group (Online Figure VIII). Consistent with the increased current amplitude, TRPM7 expression at mRNA level assessed by qPCR using mouse cardiac fibroblasts was increased by 2.8 fold. Although other Ca2+-permeable channel genes were also up-regulated by TGF-β1 (Online Figure IX), TRPM7 is the only functional channel which can be measured by current recording in human atrial fibroblasts (Figure 2-3) and in mouse fibroblasts before and after after TGF-β1 treatment (data not shown). Moreover, knocking down the abundantly expressed TRPC1 did not alter Ca2+ influx or differentiation of human atrial fibroblasts (Online Figure IV). Therefore, it is likely that TRPM7 plays a major role in mediating Ca2+ in atrial fibroblasts under normal conditions and upon TGF-β stimulation. To further understand the correlation between the changes of TRPM7 induced by TGFβ1 and fibroblast differentiation, we tested TRPM7 protein and α-SMA expression levels by using mouse cardiac fibroblasts. Figure 8C-D shows that TGF-β1 increased TRPM7 and α-SMA protein expression by 3.8- and 2.1-fold, respectively. The mRNA level of collagen (type I) as well as collagen production measured in human atrial fibroblasts was markedly increased by TGF-β1 (Fig. 8E-F). These results further suggest that up-regulation of TRPM7 underlies the Ca2+ signaling mechanism for TGF-β1 induced fibroblast differentiation and collagen production, and that TRPM7 may play a role in TGF-β1 induced atrial fibrogenesis.
Figure 8.
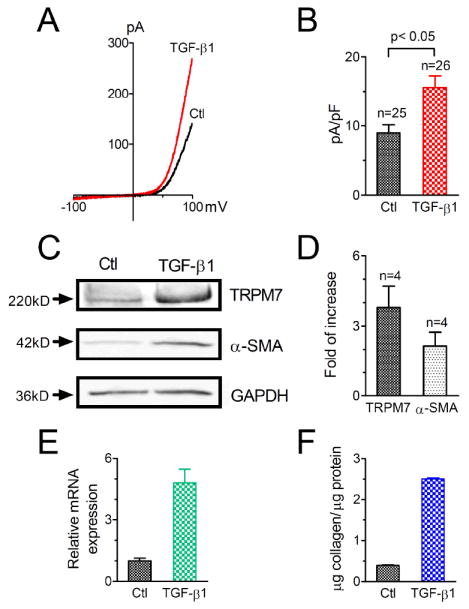
Up-regulation of TRPM7 by TGF-β1. A, Representative recording of TRPM7 in control human atrial fibroblasts and in fibroblasts treated with 10 ng/ml TGF-β1. B, Average current amplitude of TRPM7 after TGF-β1 treatment in comparison with control cells. The current density was 8.9 ± 1.2 (n=25) pA/pF in control and 15.6 ± 1.7 (n=26) pA/pF in TGF-β1 treated group. C, Up-regulation of TRPM7 by TGF-β1 evaluated in mouse cardiac fibroblasts. Top: western blot results of TRPM7 in control and TGF-β1 treated groups; Middle: expression of α-SMA in control and TGF-β1treated groups; Bottom: GAPDH used as loading control. D, Average increase in TRPM7 and α-SMA expression induced by TGF-β1. TRPM7 protein expression was increased by 3.8 ± 0.9 fold (n=4) and α-SMA was increased by 2.1 ± 0.6 fold (n=4). E-F, Increased collagen production in human atrial fibroblasts induced by TGF-β1 evaluated at mRNA (E) and protein (F) levels. Average data were from three separate experiments.
DISCUSSION
To our knowledge, this is the first study to report a pathophysiological function of the Ca2+-permeable cation channel TRPM7 in human disease. We made several new findings in this study. First, our results reveal that TRPM7 underlies the molecular mechanism of Ca2+ signaling in human atrial fibroblasts. Second, we demonstrate that TRPM7 mediated Ca2+ plays an essential role in atrial fibroblasts proliferation and differentiation. Third, we show that TRPM7 is markedly up-regulated in AF patients, and that up-regulation of TRPM7 confers increased fibroblast differentiation in AF patients. Our results suggest that TRPM7 mediated Ca2+ may play a pivotal role in fibrogenesis in human AF.
TRPM7 underlies the molecular mechanism of Ca2+ signaling in human atrial fibroblasts
Ca2+ signals are essential for various cellular functions. Although the importance of fibrosis in various cardiac diseases has been well appreciated, the Ca2+ signaling mechanisms in cardiac fibrogenesis are not known. Here we demonstrate that TRPM7 is abundantly expressed in human atrial fibroblasts, and is the major Ca2+-permeable channel which is responsible for Ca2+ influx in human atrial fibroblasts.
TRPM7 is a Ca2+-permeable cation channel 36, 37. It is constitutively active and brings Ca2+ into cells under physiological conditions. High intracellular Mg2+ concentration blocks TRPM7. However, TRPM7 channels show ~50% of their maximum activity at physiological Mg2+ levels (0.5–1 mM) 36, 51. Extracellular low pH potentiates TRPM7 inward current 42, and oxidative stress activates TRPM7 40. Membrane stretch 52 and shear stress 38 have been reported to activate TRPM7. All of these characteristics of TRPM7 suggest that TRPM7 plays a role under pathological conditions, such as ischemia or oxidative stress, which may initiate the cardiac fibrogenesis cascade.
Our results indicate that TRPM7 is encoded by TRPM7 and that TRPM7 is the major functional Ca2+-permeable channel in human atrial fibroblasts. Although TRPV2, TRPV4, TRPC1 and TRPC6 were detectable by RT-PCR, current recording results indicate that there were no functional TRPV2, TRPV4 or TRPC6 channels in the fibroblasts, making them unlikely to contribute to Ca2+ signaling in the fibroblasts. TRPV4 is sensitive to stretch 53, and can be specifically activated by 4α-PDD. We found that in both freshly isolated and cultured fibroblasts, 4α-PDD was unable to activate TRPV4-like currents (Figure 3). TRPC6 is activated by Gq-linked receptor stimulation, DAG or OAG, or by direct application of GTPγS in the pipette solution. We could not activate TRPC6-like current by LPA, which activates the EDG7 receptor 54, or by including GTPγS in the pipette solution (Online Figure III). These results indicate that TRPC6 is not functionally expressed in human atrial fibroblasts. A previous study reported that TRPC6 was activated by C-type natriuretic peptide through PLC activation or directly by OAG in rat cardiac fibroblasts 48. We do not know the reason for this discrepancy, but human and rat cardiac fibroblasts may express TRPC6 differently.
Store-depletion activated Ca2+ current (ICRAC) contributes to Ca2+ influx and plays an important role in cellular functions in a variety of cells 55, 56. In human atrial fibroblasts, we could not detect ICRAC currents, and did not find that store-depletion could induce Ca2+ influx (Online Figure V) in human atrial fibroblasts. ICRAC, TRPV2, TRPV4, TRPC1 and TRPC6 currents were not present in AF fibroblasts. Even though we found that TGF-β1 up-regulates mRNA expression of TRPM7, TRPV2, TRPC1, TRPC3, TRPC4 and TRPC6 (Online Figure IX), TRPM7 current is the only functional current recorded in the fibroblasts treated with TGF-β1, suggesting that TRPM7 is the major channel mediating Ca2+ entry in atrial fibroblasts. When TRPM7 was knocked down, Ca2+ influx was substantially decreased in TRPM7-shRNA treated atrial fibroblasts in comparison with Scr-shRNA treated atrial fibroblasts, indicating that TRPM7 is needed for Ca2+ entry in fibroblasts. Taken together, our results indicate that TRPM7 is the major Ca2+-permeable channel which is responsible for Ca2+ influx in human atrial fibroblasts.
Up-regulation of TRPM7 may underlie TGF-β1 induced fibroblast differentiation in AF patients
We found that TRPM7 is markedly up-regulated in the fibroblasts from AF patients. Both current density and Ca2+ influx mediated by TRPM7 from AF patients were significantly larger than those from NSR patients (Figure 4). Due to the limited amount of tissue and numbers of human atrial fibroblasts, we could not compare the TRPM7 expression in AF and NSR patients by measuring protein levels using Western Blot. Since the only differences between AF patients and NSR patients is the presence of atrial fibrillation versus normal sinus rhythm (Online Table I), it appears that atrial fibrillation is the only variable that correlates with the up-regulated TRPM7. To understand the causal importance of TRPM7 in AF, we investigated whether TRPM7 influences fibroblast differentiation. We found that fibroblasts isolated from AF patients are more prone to differentiation under culture conditions even without TGF-β1 treatment. This inherent tendency to differentiation could be due to the increased expression level of TRPM7 when the fibroblasts were exposed to the residual amount of TGFβ in the culture media containing serum 50. Indeed, knocking-down TRPM7 by TRPM7-shRNA not only markedly reduced basal differentiation of AF fibroblasts (Fig. 6C-D), but also substantially decreased the number of differentiated myofibroblasts and the expression level of α-SMA induced by TGF-β1, indicating that TRPM7 is necessary for fibroblast differentiation. As the myofibroblasts are the primary cells that synthesize ECM and cytokines, which in turn stimulate the fibrogenesis cascade, the requirement of TRPM7 for myofibroblast differentiation indicates that TRPM7 mediated Ca2+ signals play a pivotal role in fibrosis formation.
Fibrogenesis can be triggered by various stimuli, including mechanical stretch, oxidative stress, hormones, myocardial injury, and autocrine-paracrine mediators 14, 16, among which TGF-β1 is the most potent stimulator in both in vivo and in vitro conditions. Interestingly, we found that TRPM7 expression was significantly increased by TGF-β1 in cultured fibroblasts (Figure 8). The increased TRPM7 expression level induced by TGF-β1 parallels the fibroblast differentiation as assessed by α-SMA expression and collagen production (Figure 8C-F). These results indicate that TRPM7 is indispensable for TGF-β1 induced differentiation, and that the up-regulated TRPM7 may synergize with TGFβ1 in inducing fibroblast differentiation. It is possible that the up-regulation of TRPM7 by TGF-β1 in cultured fibroblasts represents the major mechanism by which TRPM7 is up-regulated in AF patients in vivo. Taken together, our results suggest the following model: In human atria, TGF-β1 is activated by mechanical stretch, oxidative stress, or other stimuli. TGF-β1 up-regulates TRPM7, which in turn brings more Ca2+ for TGF-β1 to induce differentiation. The differentiated myofibroblasts synthesize ECM and cytokines, and excessive accumulation of ECM forms fibrosis. Cytokines can in turn stimulate TGF-β1 and cause up-regulation of TRPM7, leading to perpetuation of the fibrogenesis process.
Potential mechanism by which Ca2+signals are involved in TGF-β1 induced fibrosis
Atrial fibrosis plays an important role in the pathology of AF. Although three interrelated pathways including the renin-angiotensin system, TGF-β1 and the oxidative stress pathways have been shown to be involved in fibrosis 57-63, many fundamental aspects of fibrogenesis are still to be established 6, 57. Our results indicate that TRPM7-mediated Ca2+ signal is another fundamental factor contributing to atrial fibrogenesis cascade.
How does the Ca2+ signal contribute to fibrosis formation? There are several possibilities. First, Ca2+-dependent but TGF-β1-independent pathways may control fibrotic gene expression 64-67. Second, Ca2+ signals may influence TGF-β1 via the Smad-independent pathway 68-70. Third, TGF-β1 can induce Ca2+ entry 71, 72 or integrate with reactive oxygen species (ROS) associated Ca2+ influx and therefore influence ECM protein synthesis 73 through a ROS activated calcineurin pathway. As TRPM7 can be activated by ROS 40, it is possible that the ROS activated calcineurin pathway may have contributed to the up-regulation of TRPM7 by TGF-β1 in human atrial fibroblasts. Fourth, TRPM7-mediated Ca2+ signals may activate the calcineurin pathway and produce synergistic effect with TGF-β1 (Smad-dependent pathway) in regulating gene expression. Further studies are required to understand how TRPM7 mediated Ca2+ signals contribute to TGF-β1 induced fibrogenesis. Nevertheless, our findings that TRPM7 is the molecular basis for the major Ca2+ entry channel in cardiac fibroblasts provide a necessary tool to unravel the mechanisms by which Ca2+ signals contribute to fibrogenesis.
Potential significance
Atrial fibrosis can be triggered by various factors 13 via different pathways 57. Blocking one pathway may not be able to attenuate or eliminate fibrosis given the complex pathological conditions. The TRPM7-mediated Ca2+ signals which are necessary for TGF-β1 induced fibrogenesis may serve as a common pathway in the fibrogenesis cascade. If so, inhibition of Ca2+ entry through TRPM7 may attenuate cardiac fibrosis regardless of the original stimuli.
TRPM7 has been shown to be essential for cell viability 74, 75 and is responsible for Ca2+ overload induced cell death under anoxia conditions 40. Although previous studies suggested that TRPM7 is involved in Mg2+ homeostasis 74, 76, knocking out of the TRPM7 gene disrupts embryonic development but does not alter Mg2+ homeostasis 39, indicating that Ca2+ permeation may confer major channel functions of TRPM7. The contribution of the TRPM7-mediated Ca2+ signal in human atrial fibrogenesis shown in this study is the first demonstration of an important role of this unique channel in human diseases.
Conclusions
In this study, we establish that the Ca2+-permeable cation channel TRPM7 underlies Ca2+ signaling mechanisms in the cardiac fibrogenesis, making TRPM7 the first known Ca2+-permeable channel involved in the genesis of cardiac fibrosis. More importantly, our results demonstrate that TRPM7 plays an essential role in fibrogenesis in human AF. Our study opens a new avenue to explore the potential roles of Ca2+ signaling in fibrogenesis, which will lead to better understanding of the mechanism of cardiac fibrogenesis, and could ultimately results in development of more effective approaches for treatment of AF.
Supplementary Material
Acknowledgments
We thank Drs Stanley Nattel, Laurinda Jaffe, Achilles Pappano, and Haoxing Xu for constructive suggestions and comments. We also thank Philip Batista, Mariela Agosto and Dorota Pawlak for help with the patient database. The authors appreciate helpful comments from other members in the lab.
SOURCES OF FUNDING This work was partially supported by National Institutes of Health Grant HL078960 (L. Y.), a bio-medical grant from the Department of Public Health of Connecticut (L. Y.), and institutional funding of UCHC (L. Y).
Non-standard Abbreviations and Acronyms
- AF
Atrial Fibrillation
- NSR
Normal sinus rhythm
- TRPM7
Transient receptor potential melastatin related 7
- TRP
Transient receptor potential
- TRPV2
Transient receptor potential V2
- TRPV4
Transient receptor potential V4
- TRPC6
Transient receptor potential C6
- CABG
Coronary Artery Bypass Graft
- AngII
Angiotensin II
- 2-APB
2-Aminoethoxydiphenyl borate
- TGF-β1
Transforming growth factor β1
- ECM
Extracellular matrix proteins
- shRNA
Small hairpin RNA
- 4α-PDD
4α-Phorbol-12,13-didecanoate
- DAG
Diaglycerol
- OAG
1-Oleoyl-2-acetyl-sn-glycerol
- α-SMA
alpha-Smooth Muscle Actin
- EDG7
a Lysophospholipid/Lysosphingolipid Receptor
- ROS
Reactive oxygen species
- Tg
Thapsigargin
- CCH
Carbachol
- LPA
Oleoyl-lysophosphatidic acid
NOVELTY AND SIGNIFICANCE
What is known?
Atrial fibrosis is a hallmark feature of structural remodeling in atrial fibrillation (AF).
Previous studies have suggested that Ca2+ entry is essential for fibroblast function. However, the Ca2+ signaling mechanisms in cardiac fibrogenesis have not elucidated.
What new information does this article contribute?
Here we report that the Ca2+-permeable cation channel TRPM7 is the major Ca2+-permeable channel in human atrial fibroblasts.
TRPM7-mediated Ca2+ entry is essential for TGF-β1-induced fibroblast differentiation.
TRPM7 is significantly up-regulated in AF fibroblasts and plays a key role in enhanced fibroblast proliferation, differentiation, and collagen production during AF.
Summary
Atrial fibrosis is a major factor contributing to AF. Understanding the mechanism of fibrogenesis is crucial for developing new therapeutic strategies for attenuating or reversing fibrosis-associated AF. The aim of this study was to elucidate the Ca2+ signaling mechanisms in fibroblasts and how Ca2+ signals contribute to fibrogenesis in AF. We discovered that TRPM7, a Ca2+-permeable cation channel that possesses protein kinase function, is the key Ca2+-permeable channel in human atrial fibroblasts. TRPM7 and TRPM7-mediated Ca2+ signals were drastically up-regulated in AF patients, which correlated with enhanced fibrogenesis in cells from these patients. This study identifies for the first time the molecular nature of the major Ca2+-permeable channel in cardiac fibroblasts, which helps to elucidate how Ca2+ signals contribute to fibrogenesis in human AF. Also, our observation that TRPM7-mediated Ca2+ entry contributes to human AF represents the first report of an important role of TRPM7 in human diseases. Our study opens a new avenue to explore the potential role of Ca2+ signals in fibrogenesis, which should lead to better understanding of the mechanisms of cardiac fibrogenesis and could ultimately result in the development of more effective approaches for the treatment of AF.
Footnotes
DISCLOSURES None.
Contributor Information
Jianyang Du, Calhoun Cardiology Center, University of Connecticut Health Center, Farmington, Connecticut 06030, USA; Department of Cell Biology, University of Connecticut Health Center, Farmington, Connecticut 06030, USA.
Jia Xie, Calhoun Cardiology Center, University of Connecticut Health Center, Farmington, Connecticut 06030, USA; Department of Cell Biology, University of Connecticut Health Center, Farmington, Connecticut 06030, USA.
Zheng Zhang, Calhoun Cardiology Center, University of Connecticut Health Center, Farmington, Connecticut 06030, USA; Department of Cell Biology, University of Connecticut Health Center, Farmington, Connecticut 06030, USA.
Hiroto Tsujikawa, Calhoun Cardiology Center, University of Connecticut Health Center, Farmington, Connecticut 06030, USA; Department of Cell Biology, University of Connecticut Health Center, Farmington, Connecticut 06030, USA.
Daniel Fusco, Calhoun Cardiology Center, University of Connecticut Health Center, Farmington, Connecticut 06030, USA.
David Silverman, Calhoun Cardiology Center, University of Connecticut Health Center, Farmington, Connecticut 06030, USA.
Bruce Liang, Calhoun Cardiology Center, University of Connecticut Health Center, Farmington, Connecticut 06030, USA.
Lixia Yue, Calhoun Cardiology Center, University of Connecticut Health Center, Farmington, Connecticut 06030, USA; Department of Cell Biology, University of Connecticut Health Center, Farmington, Connecticut 06030, USA.
References
- 1.Weber K. Cardiac interstitium. In: Poole-Wilson P, Colucci W, Massie B, Chatterjee K, Coats A, editors. Heart Failure. New York, NY: Churchill Livingstone; 1997. pp. 13–31. [Google Scholar]
- 2.Nattel S, Li D, Yue L. Basic mechanisms of atrial fibrillation--very new insights into very old ideas. Annu Rev Physiol. 2000;62:51–77. doi: 10.1146/annurev.physiol.62.1.51. [DOI] [PubMed] [Google Scholar]
- 3.Ausma J, Wijffels M, Thone F, Wouters L, Allessie M, Borgers M. Structural changes of atrial myocardium due to sustained atrial fibrillation in the goat. Circulation. 1997;96:3157–3163. doi: 10.1161/01.cir.96.9.3157. [DOI] [PubMed] [Google Scholar]
- 4.Wijffels MC, Kirchhof CJ, Dorland R, Allessie MA. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation. 1995;92:1954–1968. doi: 10.1161/01.cir.92.7.1954. [DOI] [PubMed] [Google Scholar]
- 5.Allessie M, Ausma J, Schotten U. Electrical, contractile and structural remodeling during atrial fibrillation. Cardiovasc Res. 2002;54:230–246. doi: 10.1016/s0008-6363(02)00258-4. [DOI] [PubMed] [Google Scholar]
- 6.Burstein B, Nattel S. Atrial Fibrosis: Mechanisms and Clinical Relevance in Atrial Fibrillation. Journal of the American College of Cardiology. 2008;51:802–809. doi: 10.1016/j.jacc.2007.09.064. [DOI] [PubMed] [Google Scholar]
- 7.Kostin S, Klein G, Szalay Z, Hein S, Bauer EP, Schaper J. Structural correlate of atrial fibrillation in human patients. Cardiovasc Res. 2002;54:361–379. doi: 10.1016/s0008-6363(02)00273-0. [DOI] [PubMed] [Google Scholar]
- 8.Boldt A, Wetzel U, Lauschke J, Weigl J, Gummert J, Hindricks G, Kottkamp H, Dhein S. Fibrosis in left atrial tissue of patients with atrial fibrillation with and without underlying mitral valve disease. Heart. 2004;90:400–405. doi: 10.1136/hrt.2003.015347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu J, Cui G, Esmailian F, Plunkett M, Marelli D, Ardehali A, Odim J, Laks H, Sen L. Atrial extracellular matrix remodeling and the maintenance of atrial fibrillation. Circulation. 2004;109:363–368. doi: 10.1161/01.CIR.0000109495.02213.52. [DOI] [PubMed] [Google Scholar]
- 10.Frustaci A, Chimenti C, Bellocci F, Morgante E, Russo MA, Maseri A. Histological substrate of atrial biopsies in patients with lone atrial fibrillation. Circulation. 1997;96:1180–1184. doi: 10.1161/01.cir.96.4.1180. [DOI] [PubMed] [Google Scholar]
- 11.Nakajima H, Nakajima HO, Salcher O, Dittie AS, Dembowsky K, Jing S, Field LJ. Atrial but not ventricular fibrosis in mice expressing a mutant transforming growth factor-beta(1) transgene in the heart. Circ Res. 2000;86:571–579. doi: 10.1161/01.res.86.5.571. [DOI] [PubMed] [Google Scholar]
- 12.Verheule S, Sato T, Everett Tt, Engle SK, Otten D, Rubart-von der Lohe M, Nakajima HO, Nakajima H, Field LJ, Olgin JE. Increased vulnerability to atrial fibrillation in transgenic mice with selective atrial fibrosis caused by overexpression of TGF-beta1. Circ Res. 2004;94:1458– 1465. doi: 10.1161/01.RES.0000129579.59664.9d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manabe I, Shindo T, Nagai R. Gene Expression in Fibroblasts and Fibrosis: Involvement in Cardiac Hypertrophy. Circ Res. 2002;91:1103–1113. doi: 10.1161/01.res.0000046452.67724.b8. [DOI] [PubMed] [Google Scholar]
- 14.Swynghedauw B. Molecular mechanisms of myocardial remodeling. Physiol Rev. 1999;79:215–262. doi: 10.1152/physrev.1999.79.1.215. [DOI] [PubMed] [Google Scholar]
- 15.Sun Y, Weber KT. Infarct scar: a dynamic tissue. Cardiovasc Res. 2000;46:250–256. doi: 10.1016/s0008-6363(00)00032-8. [DOI] [PubMed] [Google Scholar]
- 16.Weber KT, Sun Y, Tyagi SC, Cleutjens JP. Collagen network of the myocardium: function, structural remodeling and regulatory mechanisms. J Mol Cell Cardiol. 1994;26:279–292. doi: 10.1006/jmcc.1994.1036. [DOI] [PubMed] [Google Scholar]
- 17.Goumans MJ, van Zonneveld AJ, ten Dijke P. Transforming growth factor beta-induced endothelial-to-mesenchymal transition: a switch to cardiac fibrosis? Trends Cardiovasc Med. 2008;18:293–298. doi: 10.1016/j.tcm.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 18.Powell DW, Mifflin RC, Valentich JD, Crowe SE, Saada JI, West AB. Myofibroblasts. I. Paracrine cells important in health and disease. Am J Physiol. 1999;277:C1–9. doi: 10.1152/ajpcell.1999.277.1.C1. [DOI] [PubMed] [Google Scholar]
- 19.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 20.Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 21.Kumaran C, Shivakumar K. Calcium- and superoxide anion-mediated mitogenic action of substance P on cardiac fibroblasts. Am J Physiol Heart Circ Physiol. 2002;282:H1855–1862. doi: 10.1152/ajpheart.00747.2001. [DOI] [PubMed] [Google Scholar]
- 22.Shivakumar K, Kumaran C. L-type calcium channel blockers and EGTA enhance superoxide production in cardiac fibroblasts. J Mol Cell Cardiol. 2001;33:373–377. doi: 10.1006/jmcc.2000.1309. [DOI] [PubMed] [Google Scholar]
- 23.Colston JT, Chandrasekar B, Freeman GL. A novel peroxide-induced calcium transient regulates interleukin-6 expression in cardiac-derived fibroblasts. J Biol Chem. 2002;277:23477–23483. doi: 10.1074/jbc.M108676200. [DOI] [PubMed] [Google Scholar]
- 24.Kiseleva I, Kamkin A, Kohl P, Lab MJ. Calcium and mechanically induced potentials in fibroblasts of rat atrium. Cardiovasc Res. 1996;32:98–111. [PubMed] [Google Scholar]
- 25.Kiseleva I, Kamkin A, Pylaev A, Kondratjev D, Leiterer KP, Theres H, Wagner KD, Persson PB, Gunther J. Electrophysiological properties of mechanosensitive atrial fibroblasts from chronic infarcted rat heart. J Mol Cell Cardiol. 1998;30:1083–1093. doi: 10.1006/jmcc.1998.0673. [DOI] [PubMed] [Google Scholar]
- 26.Kamkin A, Kiseleva I, Isenberg G. Activation and inactivation of a non-selective cation conductance by local mechanical deformation of acutely isolated cardiac fibroblasts. Cardiovasc Res. 2003;57:793–803. doi: 10.1016/s0008-6363(02)00775-7. [DOI] [PubMed] [Google Scholar]
- 27.Olson ER, Shamhart PE, Naugle JE, Meszaros JG. Angiotensin II-Induced Extracellular Signal-Regulated Kinase 1/2 Activation Is Mediated by Protein Kinase C{delta} and Intracellular Calcium in Adult Rat Cardiac Fibroblasts. Hypertension. 2008;51:704–711. doi: 10.1161/HYPERTENSIONAHA.107.098459. [DOI] [PubMed] [Google Scholar]
- 28.Ramires FJ, Sun Y, Weber KT. Myocardial fibrosis associated with aldosterone or angiotensin II administration: attenuation by calcium channel blockade. J Mol Cell Cardiol. 1998;30:475–483. doi: 10.1006/jmcc.1997.0612. [DOI] [PubMed] [Google Scholar]
- 29.Kiseleva IS, Kamkin AG, Kirkheis R, Kositskii GI. Intercellular electrotonic interactions in the cardiac sinus node in the frog. Dokl Akad Nauk SSSR. 1987;292:1502–1505. [PubMed] [Google Scholar]
- 30.Kamkin A, Kiseleva I, Wagner KD, Pylaev A, Leiterer KP, Theres H, Scholz H, Gunther J, Isenberg G. A possible role for atrial fibroblasts in postinfarction bradycardia. Am J Physiol Heart Circ Physiol. 2002;282:H842–849. doi: 10.1152/ajpheart.00240.2001. [DOI] [PubMed] [Google Scholar]
- 31.Kamkin A, Kiseleva I, Wagner KD, Lammerich A, Bohm J, Persson PB, Gunther J. Mechanically induced potentials in fibroblasts from human right atrium. Exp Physiol. 1999;84:347–356. [PubMed] [Google Scholar]
- 32.Kohl P, N D. Mechanosensitive connective tissue: potential influence on heart rhythm. Cardiovascular Research. 1996;32:62–68. [PubMed] [Google Scholar]
- 33.Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- 34.Montell C. The TRP superfamily of cation channels. Sci STKE. 2005;2005:1–24. doi: 10.1126/stke.2722005re3. [DOI] [PubMed] [Google Scholar]
- 35.Nilius B. TRP channels in disease. Biochim Biophys Acta. 2007;1772:805–812. doi: 10.1016/j.bbadis.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 36.Nadler MJ, Hermosura MC, Inabe K, Perraud AL, Zhu Q, Stokes AJ, Kurosaki T, Kinet JP, Penner R, Scharenberg AM, Fleig A. LTRPC7 is a Mg.ATP-regulated divalent cation channel required for cell viability. Nature. 2001;411:590–595. doi: 10.1038/35079092. [DOI] [PubMed] [Google Scholar]
- 37.Runnels LW, Yue L, Clapham DE. TRP-PLIK, a bifunctional protein with kinase and ion channel activities. Science. 2001;291:1043–1047. doi: 10.1126/science.1058519. [DOI] [PubMed] [Google Scholar]
- 38.Oancea E, Wolfe JT, Clapham DE. Functional TRPM7 Channels Accumulate at the Plasma Membrane in Response to Fluid Flow. Circ Res. 2006;98:245–253. doi: 10.1161/01.RES.0000200179.29375.cc. [DOI] [PubMed] [Google Scholar]
- 39.Jin J, Desai BN, Navarro B, Donovan A, Andrews NC, Clapham DE. Deletion of Trpm7 disrupts embryonic development and thymopoiesis without altering Mg2+ homeostasis. Science. 2008;322:756–760. doi: 10.1126/science.1163493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aarts M, Iihara K, Wei WL, Xiong ZG, Arundine M, Cerwinski W, MacDonald JF, Tymianski M. A key role for TRPM7 channels in anoxic neuronal death. Cell. 2003;115:863–877. doi: 10.1016/s0092-8674(03)01017-1. [DOI] [PubMed] [Google Scholar]
- 41.Yue L, Melnyk P, Gaspo R, Wang Z, Nattel S. Molecular mechanisms underlying ionic remodeling in a dog model of atrial fibrillation. Circ Res. 1999;84:776–784. doi: 10.1161/01.res.84.7.776. [DOI] [PubMed] [Google Scholar]
- 42.Jiang J, Li M, Yue L. Potentiation of TRPM7 Inward Currents by Protons. J Gen Physiol. 2005;126:137–150. doi: 10.1085/jgp.200409185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li M, Jiang J, Yue L. Functional Characterization of Homo- and Heteromeric Channel Kinases TRPM6 and TRPM7. J Gen Physiol. 2006;127:525–537. doi: 10.1085/jgp.200609502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li M, Du J, Jiang J, Ratzan W, Su L-T, Runnels LW, Yue L. Molecular Determinants of Mg2+ and Ca2+ Permeability and pH Sensitivity in TRPM6 and TRPM7. J Biol Chem. 2007;282:25817–25830. doi: 10.1074/jbc.M608972200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hu HZ, Gu Q, Wang C, Colton CK, Tang J, Kinoshita-Kawada M, Lee LY, Wood JD, Zhu MX. 2-aminoethoxydiphenyl borate is a common activator of TRPV1, TRPV2, and TRPV3. J Biol Chem. 2004;279:35741–35748. doi: 10.1074/jbc.M404164200. [DOI] [PubMed] [Google Scholar]
- 46.Watanabe H, Davis JB, Smart D, Jerman JC, Smith GD, Hayes P, Vriens J, Cairns W, Wissenbach U, Prenen J, Flockerzi V, Droogmans G, Benham CD, Nilius B. Activation of TRPV4 channels (hVRL-2/mTRP12) by phorbol derivatives. J Biol Chem. 2002;277:13569–13577. doi: 10.1074/jbc.M200062200. [DOI] [PubMed] [Google Scholar]
- 47.Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 1999;397:259–263. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- 48.Rose RA, Hatano N, Ohya S, Imaizumi Y, Giles WR. C-type natriuretic peptide activates a non-selective cation current in acutely isolated rat cardiac fibroblasts via natriuretic peptide C receptor-mediated signalling. J Physiol. 2007;580:255–274. doi: 10.1113/jphysiol.2006.120832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Strubing C, Krapivinsky G, Krapivinsky L, Clapham DE. Formation of novel TRPC channels by complex subunit interactions in embryonic brain. J Biol Chem. 2003;278:39014–39019. doi: 10.1074/jbc.M306705200. [DOI] [PubMed] [Google Scholar]
- 50.Kropf J, Schurek JO, Wollner A, Gressner AM. Immunological measurement of transforming growth factor-beta 1 (TGF-ß1) in blood; assay development and comparison. Clin Chem. 1997;43:1965–1974. [PubMed] [Google Scholar]
- 51.Kozak JA, Cahalan MD. MIC channels are inhibited by internal divalent cations but not ATP. Biophys J. 2003;84:922–927. doi: 10.1016/S0006-3495(03)74909-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Numata T, Shimizu T, Okada Y. TRPM7 is a stretch- and swelling-activated cation channel involved in volume regulation in human epithelial cells. Am J Physiol Cell Physiol. 2007;292:C460–467. doi: 10.1152/ajpcell.00367.2006. [DOI] [PubMed] [Google Scholar]
- 53.Suzuki M, Mizuno A, Kodaira K, Imai M. Impaired pressure sensation in mice lacking TRPV4. J Biol Chem. 2003;278:22664–22668. doi: 10.1074/jbc.M302561200. [DOI] [PubMed] [Google Scholar]
- 54.Runnels LW, Yue L, Clapham DE. The TRPM7 channel is inactivated by PIP(2) hydrolysis. Nat Cell Biol. 2002;4:329–336. doi: 10.1038/ncb781. [DOI] [PubMed] [Google Scholar]
- 55.Lewis RS. Calcium signaling mechanisms in T lymphocytes. Annu Rev Immunol. 2001;19:497–521. doi: 10.1146/annurev.immunol.19.1.497. [DOI] [PubMed] [Google Scholar]
- 56.Lewis RS. The molecular choreography of a store-operated calcium channel. Nature. 2007;446:284–287. doi: 10.1038/nature05637. [DOI] [PubMed] [Google Scholar]
- 57.Everett THt, Olgin JE. Atrial fibrosis and the mechanisms of atrial fibrillation. Heart Rhythm. 2007;4:S24–27. doi: 10.1016/j.hrthm.2006.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weber KT, Brilla CG, Janicki JS. Myocardial fibrosis: functional significance and regulatory factors. Cardiovasc Res. 1993;27:341–348. doi: 10.1093/cvr/27.3.341. [DOI] [PubMed] [Google Scholar]
- 59.Hanatani A, Yoshiyama M, Kim S, Omura T, Toda I, Akioka K, Teragaki M, Takeuchi K, Iwao H, Takeda T. Inhibition by angiotensin II type 1 receptor antagonist of cardiac phenotypic modulation after myocardial infarction. J Mol Cell Cardiol. 1995;27:1905–1914. doi: 10.1016/0022-2828(95)90013-6. [DOI] [PubMed] [Google Scholar]
- 60.Urata H, Boehm KD, Philip A, Kinoshita A, Gabrovsek J, Bumpus FM, Husain A. Cellular localization and regional distribution of an angiotensin II-forming chymase in the heart. J Clin Invest. 1993;91:1269–1281. doi: 10.1172/JCI116325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boldt A, Scholl A, Garbade J, Resetar ME, Mohr FW, Gummert JF, Dhein S. ACE-inhibitor treatment attenuates atrial structural remodeling in patients with lone chronic atrial fibrillation. Basic Res Cardiol. 2006;101:261–267. doi: 10.1007/s00395-005-0571-2. [DOI] [PubMed] [Google Scholar]
- 62.Hirayama Y, Atarashi H, Kobayashi Y, Horie T, Iwasaki Y, Maruyama M, Miyauchi Y, Ohara T, Yashima M, Takano T. Angiotensin-converting enzyme inhibitor therapy inhibits the progression from paroxysmal atrial fibrillation to chronic atrial fibrillation. Circ J. 2005;69:671–676. doi: 10.1253/circj.69.671. [DOI] [PubMed] [Google Scholar]
- 63.Zaman AG, Kearney MT, Schecter C, Worthley SG, Nolan J. Angiotensin-converting enzyme inhibitors as adjunctive therapy in patients with persistent atrial fibrillation. Am Heart J. 2004;147:823–827. doi: 10.1016/j.ahj.2003.07.027. [DOI] [PubMed] [Google Scholar]
- 64.Fantozzi I, Zhang S, Platoshyn O, Remillard CV, Cowling RR, Yuan JX. Hypoxia increases AP-1 binding activity by enhancing capacitative Ca2+ entry in human pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2003;285:L1233–1245. doi: 10.1152/ajplung.00445.2002. [DOI] [PubMed] [Google Scholar]
- 65.Zhang S, Fantozzi I, Tigno DD, Yi ES, Platoshyn O, Thistlethwaite PA, Kriett JM, Yung G, Rubin LJ, Yuan JX. Bone morphogenetic proteins induce apoptosis in human pulmonary vascular smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2003;285:L740–754. doi: 10.1152/ajplung.00284.2002. [DOI] [PubMed] [Google Scholar]
- 66.Biswas G, Anandatheerthavarada HK, Zaidi M, Avadhani NG. Mitochondria to nucleus stress signaling: a distinctive mechanism of NFkappaB/Rel activation through calcineurin-mediated inactivation of IkappaBbeta. J Cell Biol. 2003;161:507–519. doi: 10.1083/jcb.200211104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Apati A, Janossy J, Brozik A, Bauer PI, Magocsi M. Calcium induces cell survival and proliferation through the activation of the MAPK pathway in a human hormone-dependent leukemia cell line, TF-1. J Biol Chem. 2003;278:9235–9243. doi: 10.1074/jbc.m205528200. [DOI] [PubMed] [Google Scholar]
- 68.Funaba M, Zimmerman CM, Mathews LS. Modulation of Smad2-mediated signaling by extracellular signal-regulated kinase. J Biol Chem. 2002;277:41361–41368. doi: 10.1074/jbc.M204597200. [DOI] [PubMed] [Google Scholar]
- 69.Scherer A, Graff JM. Calmodulin differentially modulates Smad1 and Smad2 signaling. J Biol Chem. 2000;275:41430–41438. doi: 10.1074/jbc.M005727200. [DOI] [PubMed] [Google Scholar]
- 70.Zimmerman CM, Kariapper MS, Mathews LS. Smad proteins physically interact with calmodulin. J Biol Chem. 1998;273:677–680. doi: 10.1074/jbc.273.2.677. [DOI] [PubMed] [Google Scholar]
- 71.Nesti LJ, Caterson EJ, Li WJ, Chang R, McCann TD, Hoek JB, Tuan RS. TGF-beta1 calcium signaling in osteoblasts. J Cell Biochem. 2007;101:348–359. doi: 10.1002/jcb.21180. [DOI] [PubMed] [Google Scholar]
- 72.McGowan TA, Madesh M, Zhu Y, Wang L, Russo M, Deelman L, Henning R, Joseph S, Hajnoczky G, Sharma K. TGF-beta-induced Ca(2+) influx involves the type III IP(3) receptor and regulates actin cytoskeleton. Am J Physiol Renal Physiol. 2002;282:F910–920. doi: 10.1152/ajprenal.00252.2001. [DOI] [PubMed] [Google Scholar]
- 73.Gooch JL, Gorin Y, Zhang B-X, Abboud HE. Involvement of Calcineurin in Transforming Growth Factor-{beta}-mediated Regulation of Extracellular Matrix Accumulation. J Biol Chem. 2004;279:15561–15570. doi: 10.1074/jbc.M308759200. [DOI] [PubMed] [Google Scholar]
- 74.Schmitz C, Perraud AL, Johnson CO, Inabe K, Smith MK, Penner R, Kurosaki T, Fleig A, Scharenberg AM. Regulation of vertebrate cellular Mg2+ homeostasis by TRPM7. Cell. 2003;114:191–200. doi: 10.1016/s0092-8674(03)00556-7. [DOI] [PubMed] [Google Scholar]
- 75.Hanano T, Hara Y, Shi J, Morita H, Umebayashi C, Mori E, Sumimoto H, Ito Y, Mori Y, Inoue R. Involvement of TRPM7 in cell growth as a spontaneously activated Ca2+ entry pathway in human retinoblastoma cells. J Pharmacol Sci. 2004;95:403–419. doi: 10.1254/jphs.fp0040273. [DOI] [PubMed] [Google Scholar]
- 76.Sontia B, Touyz RM. Magnesium transport in hypertension. Pathophysiology. 2007;14:205–211. doi: 10.1016/j.pathophys.2007.09.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.