

Abstract
ATP-gated P2X receptors are trimeric ion channels, as recently confirmed by X-ray crystallography. However, the structure was solved without ATP and even though extracellular intersubunit cavities surrounded by conserved amino acid residues previously shown to be important for ATP function were proposed to house ATP, the localization of the ATP sites remains elusive. Here we localize the ATP-binding sites by creating, through a proximity-dependent “tethering” reaction, covalent bonds between a synthesized ATP-derived thiol-reactive P2X2 agonist (NCS-ATP) and single cysteine mutants engineered in the putative binding cavities of the P2X2 receptor. By combining whole-cell and single-channel recordings, we report that NCS-ATP covalently and specifically labels two previously unidentified positions N140 and L186 from two adjacent subunits separated by about 18 Å in a P2X2 closed state homology model, suggesting the existence of at least two binding modes. Tethering reaction at both positions primes subsequent agonist binding, yet with distinct functional consequences. Labeling of one position impedes subsequent ATP function, which results in inefficient gating, whereas tethering of the other position, although failing to produce gating by itself, enhances subsequent ATP function. Our results thus define a large and dynamic intersubunit ATP-binding pocket and suggest that receptors trapped in covalently agonist-bound states differ in their ability to gate the ion channel.
Keywords: affinity labeling, chemical modification, purinergic receptor
P2X receptors are oligomeric ATP-gated ion channels selective to cations (1) and are involved in physiological processes as diverse as synaptic transmission, the response to inflammation, and pain perception (2). Upon ATP binding, structural rearrangements of the subunit interface (3–5) lead to the opening of the ion channel (6–8), but the entire molecular sequence of events that couple ATP binding to channel opening remains unknown. The recent X-ray structure of the P2X4 receptor in a closed resting state represents in this regard a decisive step (9). It confirms the trimeric stoichiometry of the ion channel, in agreement with the fact that there are three activatable ATP-binding sites (10), and provides a structural context to interpret functional data (11).
Early studies based on mutagenesis data have identified highly conserved extracellular residues important for ATP function and have proposed that ATP binding occurs through the extracellular domain (12–19), presumably at the subunit interface (15, 17). When mapped on the crystal structure, most of these residues are observed to line a large and deep intersubunit cavity shaped like an open “jaw” and located approximately 45 Å away from the ion channel domain (9). This observation thus suggests that these residues participate in ATP binding; however, the crystal structure was solved in the absence of ATP, and therefore no direct evidence support this hypothesis to date.
We used the proximity-dependent “tethering” approach (20) to localize definitively the ATP-binding site in the P2X2 receptor. This approach has proven to be broadly applicable to other ligand-gated ion channels to probe successfully agonist- (21), antagonist- (22, 23), allosteric modulator- (24), and channel blocker-binding sites (25). It consists in a proximity-accelerated chemical reaction between a thiol-reactive ATP analogue and a single free cysteine mutant engineered in the putative ATP-binding site (Fig. 1A). We synthesized the sulfhydryl-reactive 8-thiocyano-ATP (NCS-ATP, Fig. 1B), in which the electrophilic group appended to the ATP core in position 8 of the adenine ring was determined to react selectively and irreversibly with a native cysteine residue in Na,K-ATPases and kinases (26). During reversible binding periods and due to the affinity offered by the ATP moiety, the small size of the reactive NCS group guaranties its location in the binding site. Provided that the thiol group of the substituted cysteine is correctly orientated and spatially close to the sulfur atom of the NCS group, a mixed disulfide bond is likely to be formed between the receptor and the probe, with liberation of a cyanide ion (26). This formed disulfide bond may be cleaved by adding a reducing agent, thus allowing restoration of the initial cysteine mutant (Fig. 1A), as shown previously (26). Using this affinity labeling strategy combined with patch-clamp electrophysiology, we present evidence that NCS-ATP is a P2X2 agonist that covalently labeled two previously unidentified residues N140 and L186 mutated into cysteines from two adjacent subunits separated by about 18 Å in a closed state model of the rat P2X2 receptor. We found that, depending on the tethered position, the receptors were trapped in covalently agonist-bound states that dramatically differ in their ability to gate the ion channel. Our data provide direct evidence of the intersubunit location of the ATP sites and give previously undescribed molecular insights into the ligand-receptor recognition mechanism in the P2X2 receptor.
Fig. 1.

Strategy used to identify the ATP site in the P2X receptor. (A) Schematic representation of the strategy. (B) Chemical structure of NCS-ATP; 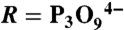 . (C) Dose-response curves from cells expressing WT P2X2 receptor (EC50 = 38 ± 3 μM, nH = 2.6 ± 0.1, Imax = 1.0, N = 7 for ATP; EC50 = 132 ± 16 μM, nH = 1.6 ± 0.2, Imax = 1.4 ± 0.2, N = 7 for NCS-ATP). (Inset) Representative traces of ATP- (100 μM) and NCS-ATP-evoked currents (1 mM) recorded from the same cell. (D) Positions mutated into single cysteine are highlighted in yellow on the three-dimensional model of the P2X2 receptor. Each subunit is depicted in a different color. Also indicated is the sequence alignment of the investigated segments (I to VI) of rat (r) and zebrafish (zf) P2X1-P2X4 and P2X7. Conserved residues previously identified as important for ATP function are underscored (12–19). Pooled data in this and all other figures represent mean ± SEM.
. (C) Dose-response curves from cells expressing WT P2X2 receptor (EC50 = 38 ± 3 μM, nH = 2.6 ± 0.1, Imax = 1.0, N = 7 for ATP; EC50 = 132 ± 16 μM, nH = 1.6 ± 0.2, Imax = 1.4 ± 0.2, N = 7 for NCS-ATP). (Inset) Representative traces of ATP- (100 μM) and NCS-ATP-evoked currents (1 mM) recorded from the same cell. (D) Positions mutated into single cysteine are highlighted in yellow on the three-dimensional model of the P2X2 receptor. Each subunit is depicted in a different color. Also indicated is the sequence alignment of the investigated segments (I to VI) of rat (r) and zebrafish (zf) P2X1-P2X4 and P2X7. Conserved residues previously identified as important for ATP function are underscored (12–19). Pooled data in this and all other figures represent mean ± SEM.
Results
Design of the Tools and of the Reaction Protocol.
The NCS-ATP was synthesized in pure form according to an adapted procedure (26) (Fig. S1A) and evoked robust, stable, and reversible inward currents upon washing in HEK-293 cells expressing the WT rat P2X2 receptor as monitored by whole-cell patch-clamp electrophysiology (Fig. 1C, Inset). Dose-response relationships showed that NCS-ATP displayed lower potency but higher efficacy than ATP (Fig. 1C). The probe was stable in buffers (< 4% degradation in 4 h) and reacted selectively and rapidly (in less than 1 min) with N-acetyl-L-cysteine methyl ester as previously described (26) (Fig. S1B). Thus, the agonist property of NCS-ATP along with its stability in water and exquisite reactivity toward cysteine residue prompted us to further use this ligand as a thiol-reactive agonist to trap engineered ATP sites in the P2X2 receptors.
On the basis of our P2X2 homology model (5), we individually mutated into cysteine 26 positions that protrude in the putative binding cavity, including those previously identified conserved residues (12–19) (Fig. 1D). All cysteine mutants, except for K69C, K71C, and K308C, were functional (Table S1).
The reactivity was monitored by systematically exposing each receptor mutant to fixed NCS-ATP reaction conditions (30 μM for 2 min). After a 5-min wash, ATP responses were recorded and compared to those measured before treatment (Fig. 2A). Remarkably, only the N140C mutant displayed robust and irreversible reduction of ATP responses (Fig. 2B). As expected, exposure to the reducing agent DTT restored ATP responses and no irreversible inhibition was observed when ATP, instead of NCS-ATP, was used (Fig. 2 and Fig. S2 A and B). On the other hand, the reversible inhibitions observed in WT, E84C, and Q138C mutants that showed little sensitivity to DTT are likely due to desensitization following continued application of agonist.
Fig. 2.

NCS-ATP covalently labels N140C and L186C mutants. (A) Representative traces showing the screening protocol. ATP-evoked currents through WT P2X2 (30 μM), N140C (100 μM), and L186C (1 mM) before and after exposure to NCS-ATP (30 μM, 2 min indicated by the red arrowhead), followed by 5-min wash (gray arrow), and 10-s DTT application (10 mM, blue arrowhead). (B) Pooled data for each cysteine mutant and WT P2X2 receptors (N = 3–8); nr, no response; na, not applicable because of no NCS-ATP effect (effects were considered when change of |ATP currents| was > ± 0.5). Arrowheads indicate labeled residues.
Surprisingly, a potentiation of ATP responses was observed for L186C mutant following the application of NCS-ATP, even after extensive washout (Fig. 2). Exposure to 10 s DTT did not restore ATP responses; however, increasing the time exposure to DDT up to 30 s almost completely restored ATP responses (Fig. S2D). Control experiments performed with ATP further confirmed that the irreversible functional effect is owed to the presence of the thiol-reactive group (Fig. S2 C and D). Overall, these results strongly suggest that N140C and L186C mutants are labeled with NCS-ATP.
To confirm that these mutants are water-accessible we used thiol-reactive methanethiosulfonate (MTS) derivatives and showed that these compounds profoundly and rapidly modified ATP responses in N140C and L186C mutants, but not in the WT receptor, showing that the side chains of N140 and L186 are quite accessible to solvent, a fact that is consistent with a water-exposed binding pocket (Fig. S3). Interestingly, the positively charged 2-aminoethyl MTS hydrobromide (MTSEA) potentiated ATP responses in L186C mutant, whereas the negatively charged sodium (2-sulfonatoethyl) MTS (MTSES) abolished responses (Fig. S3C), suggesting a charge-dependent modification.
NCS-ATP Targets the ATP-Binding Sites.
Although NCS-ATP shares high structural similarity with ATP, which strongly argues for a common binding site for the two agonists, we were still concerned about the possibility that the probe might target sites that are not relevant to ATP. In the following, we present four lines of evidence that NCS-ATP specifically targets the ATP-binding site.
First, if modification arises from affinity labeling as suggested in Fig. 1A, then the apparent tethering reaction rates kapp should be limited to NCS-ATP occupancy and thereof to its concentration. As expected, we found that kapp values saturated with increasing NCS-ATP concentration for both N140C (Fig. 3A) and L186C mutants (Fig. 4A).
Fig. 3.

NCS-ATP targets the ATP-binding site at N140C mutant. (A, Left and Middle) Kinetics of inhibition by NCS-ATP (100 μM in the representative trace or indicated concentrations in μM) or by 200 μM NCS-Ado. (Right) kapp plotted versus NCS-ATP concentrations. Data were fitted with Eq. 1 (KD = 36 ± 13 μM, kmodification = 0.09 ± 0.01 s-1, N = 5–6; see Materials and Methods). (B) Inhibition by NCS-ATP (100 μM, 2 min) in the absence or presence of 100 μM TNP-ATP (N = 8). (C) ATP dose-response curves before (EC50 = 92 ± 11 μM, nH = 2.4 ± 0.4, N = 5) and after (EC50 = 957 ± 123 μM, nH = 1.0 ± 0.1, N = 5) treatment by 30 μM NCS-ATP (2 min). (D) Inhibition (41 ± 14% of control, N = 6 outside-out patches) by NCS-ATP (100 μM, 10 s) of NPo of channels activated by ATP (10 μM, 2 s, indicated by the white bar, openings downward). Channel unitary conductance before and after NCS-ATP treatment is 22.8 ± 2.2 and 25.5 ± 2.8 pS, respectively (N = 6 patches). (E) Docking of NCS-ATP in the P2X2 model displayed in surface (Left) and cartoon (Right) representations, in which N140 is mutated into cysteine (indicated in yellow). Residues previously shown to be important for ATP function (12–19) as well as the head domain that protrudes from the binding site are also shown.
Fig. 4.

NCS-ATP targets the ATP-binding site at L186C mutant. (A, Left) Kinetics of potentiation by NCS-ATP (200 μM in the representative trace). (Right) kapp plotted versus NCS-ATP or NCS-Ado concentrations. NCS-ATP data were fitted with Eq. 1 (KD = 101 ± 26 μM and kmodification = 3.1 ± 0.4 s-1, N = 4–7). (B) Potentiation by NCS-ATP (30 μM) in the absence or presence of 300 μM TNP-ATP or 3 mM ATP (N = 4–5). (C) ATP dose-response curves before (EC50 = 1,233 ± 107 μM, nH = 1.9 ± 0.1, N = 7) and after (EC50 = 445 ± 183 μM, nH = 1.0 ± 0.2, N = 4) treatment by 30 μM NCS-ATP (20 s). (D) Potentiation (604 ± 164% of control, N = 7 patches) by NCS-ATP (10 μM, 10 s) of NPo of channels activated by ATP (30 μM, 2 s). Channel unitary conductance before and after NCS-ATP treatment is 25.1 ± 2.1 and 24.7 ± 2.3 pS, respectively (N = 7 patches). (E) Docking of NCS-ATP in the P2X2 model displayed in surface (Left) and cartoon (Right) representations, in which L186 is mutated into cysteine (indicated in yellow). Residues previously shown to be important for ATP function (12–19) as well as the head domain that protrudes from the binding site are also shown.
Second, it is known that adenosine (Ado) is a poor P2X ligand (27), and consequently its site occupancy should be lower than that of ATP. Therefore, the apparent rates of the tethering reaction by NCS-Ado should be slower than those determined by NCS-ATP. We thus synthesized NCS-Ado (Fig. S1A), verified that it was not an agonist and had no effect on ATP response in the WT P2X2 receptor, and showed that, as expected, the tethering reactions occurred for both mutants much more slowly than those produced by NCS-ATP at the same concentration (Figs. 3A and 4A). In L186C mutant, tethered NCS-Ado, observed at high concentration, also resulted in a potentiation of ATP response, suggesting that the adenosine moiety is sufficient to mediate the potentiation effect (Fig. S4).
Third, the tethering reactions observed for both mutants were largely reduced or prevented by the presence of a saturating concentration of ATP or of the competitive P2X2 antagonist 2′,3′-O-(2,4,6-trinitrophenyl)-ATP (28) (TNP-ATP), indicating pharmacological protection (Figs. 3B and 4B). Interestingly, a concentration of TNP-ATP that abolished ATP response in L186C slowed rates of NCS-ATP-induced potentiation, but did not abolish it (Fig. 4B), suggesting that the ability of the antagonist TNP-ATP to prevent the tethering reaction in L186C is not as strong as that of ATP.
Fourth, in support of the results obtained from the MTS derivatives, we found that negatively charged mutations largely decreased ATP potency (N140D and L186C, for which the side chain may be partially negatively charged at pH 7.3) or even abolished ATP function (L186E mutant, for which we verified that cell-surface expression appeared normal compared to WT, Fig. S5), suggesting that repulsive electrostatic interactions occur between the negatively charged mutant residues and the negatively charged ATP (Tables S1 and S2). By contrast, aromatic mutations (N140W and L186F) increased by about 10 times ATP potency, a fact that suggests a possible π-π interaction between the substituted aromatic residues and the adenine ring of ATP (Tables S1 and S2).
We constructed models of the cysteine mutants, which likely represent the closed state of the receptor (8, 9), to dock NCS-ATP. We found two energetically favorable poses that fit with the experimental data, in which the NCS moiety is located closely (between 4.3 to 4.6 Å) to the sulfhydryl groups of N140C (Fig. 3E) and L186C (Fig. 4E) from two adjacent subunits. Of note, the residues previously found to be important for ATP function (12–19) are in close proximity to docked NCS-ATP. Our results thus demonstrate that the intersubunit cavities found in the X-ray structure (9) are the ATP-binding sites.
The ATP-Binding Sites Are Trapped in Covalently Agonist-Bound States That Differ in Their Ability to Gate the Channel.
NCS-ATP labeled two cysteine mutants, N140C and L186C, that are separated by about 18 Å in the closed state of the receptor (Figs. 3E and 4E). Yet a crucial difference should be noted; whereas the irreversible attachment at N140C resulted in a decrease of both ATP efficacy and nominal open probability (NPo) of channels activated by ATP in outside-out patches (Fig. 3 C and D), locking at L186C trapped the receptor in a state that is more sensitive to ATP with a concomitant increase of NPo (Fig. 4 C and D), both without changing the single-channel conductance. Importantly, both mutants labeled covalently by NCS-ATP did not produce spontaneous channel gating following washout of patches (Fig. S6), a fact that confirms the absence of irreversible sustained inward currents following washout of cells (Figs. 3A and 4A). Finally, the unitary conductance of channels activated by NCS-ATP was similar to that activated by ATP, both in mutant and WT receptors (Fig. S7). Overall, these findings suggest that (i) the covalent attachment of the agonist does not affect ion permeation, (ii) attached ATP by itself is not sufficient to gate the channel, and (iii) the ATP-binding sites were trapped in covalently agonist-bound states that differ in their ability to gate the channel.
Discussion and Conclusion
In this paper, we identify the ATP-binding sites in the P2X2 receptor using a strategy that combines cysteine scanning mutagenesis and affinity labeling (20). We synthesized the affinity marker NCS-ATP and showed that the probe is an agonist on P2X2 receptor. We present evidence that NCS-ATP covalently and specifically labels N140C and L186C mutants. These two positions contribute to the ATP site, and according to the closed state model of a P2X2 receptor these residues belong to two adjacent subunits and are separated by approximately 18 Å, a distance that is compatible with the length of ATP in an extended conformation (Figs. 3E and 4E). This suggests that ATP spans the site with two distinct orientations and/or the structure of the site changes dramatically upon ATP binding. Interestingly, N140 residue is located in the “head” domain that delineates part of the binding site jaw (Figs. 3E and 4E), a result that highlights the implication of this domain in agonist binding.
The occurrence of affinity labeling is determined by important geometrical factors such as the distance between the nucleophilic sulfhydryl function of the cysteine residue and the electrophilic NCS moiety of ATP, and also by the time spent by the affinity marker in the binding site (that is correlated to the binding affinity). The fact that only two cysteine mutants out of 26 are successfully labeled, in which some have been shown to be readily modified by MTS derivates (12, 18) suggests therefore that the reaction not only depends on the intrinsic chemical reactivity of the cysteine, but is also highly specific to the cysteine-substituted position and thus to the poses that NCS-ATP adopts in the binding pocket.
According to the affinity labeling theory (20), the modification should be limited by the site occupancy of the affinity marker and also should be altered by prior addition of a saturating concentration of agonist or competitive antagonist. We indeed present evidence that the labeling reaction saturates with increasing NCS-ATP concentrations and is pharmacologically prevented by ATP or by the competitive antagonist TNP-ATP. In addition, we found that the affinity labeling is dependent on the presence of the ATP phosphate tail, because contrary to NCS-ATP no substantial labeling arises at low concentrations of NCS-Ado, a poor P2X ligand (27). Finally, we showed that modification of the side chain at N140 and L186 positions either genetically by site-directed mutagenesis or chemically by MTS derivatives of substituted cysteine profoundly alters ATP function. Overall, these data strongly suggest that (i) the residues labeled by NCS-ATP contribute to the ATP sites and (ii) the site remains capable of binding NCS-ATP even though cysteine mutations have decreased potency or sensitivity for ATP. Therefore, these data demonstrate that the intersubunit cavities found in the X-ray structure (9) are the ATP-binding sites.
Interestingly, we noted that TNP-ATP only slows down the tethering reaction for L186C mutant, whereas ATP abolishes it. This could be explained by differences in the rates of binding (kon) and/or unbinding (koff) between ATP and TNP-ATP, which are actually unknown, and/or by the fact that even though both ligands compete for the same pharmacological site, the tethering reaction may occur in a state that is preferentially reached by agonists, but not by antagonists such as TNP-ATP.
Because NCS-ATP is covalently attached to the ATP-binding site and the reaction is kinetically completed, how can the receptor continue to respond to subsequent ATP application? A first hypothesis is that the tethered agonist cannot totally occupy the binding site, and consequently free ATP molecule can still bind in the labeled pocket. We cannot formally rule out this possibility, but this seems unlikely because this would require that two ATP molecules bind simultaneously in a restricted area (approximately 18 Å), a situation that should be energetically unfavorable because of the presence of the negatively charged phosphate groups that could produce strong electrostatic repulsion. An alternative hypothesis is that attached ATP partially traps one (or two) of the three ATP-binding sites per receptor leaving the remaining unreacted site(s) available for subsequent ATP binding. This idea is consistent with our experiments, in which the slope of the dose-response relationship (nH) was shallower after completed tethering (Figs. 3C and 4C). In this context, we propose that the tethering reaction induces an allosteric change that primes agonist binding at neighboring site(s) and concomitantly modifies the accessibility of the unreacted cysteine(s) that in turn restricts the reaction to only one or two site(s). The precise mechanism underlying the tethering stoichiometry deserves to be further addressed by additional experiments.
Based on the aforementioned hypothesis that the receptor is partially and covalently liganded by NCS-ATP, we were surprised to find that this covalently agonist-bound state in the L186C mutant fails to lock the channel in the open state. Two explanations can be proposed. First, attached ATP does not fit exquisitely within the site that results in unproductive binding. However, this is inconsistent with the fact that the tethering reaction results in enhanced subsequent ATP function. A second explanation is that a partially liganded state cannot produce significant gating, as previously observed in a single-channel study performed in P2X2 receptor, which showed that channels only open after being fully liganded (10). This appealing hypothesis also deserves to be further explored, but finds precedents in the cyclic nucleotide-gated channels, in which monoliganded channels open with a very low probability that is not different from spontaneous channel opening (29).
In conclusion, the present study provides direct evidence that the intersubunit cavities found in the X-ray structure represent the ATP-binding sites in the P2X2 receptor. We found that the adenine base of the NCS-ATP molecule is close to two previously unidentified residues N140 and L186 and that interaction of these residues with ATP may contribute to its binding energy. Our results suggest that receptors trapped in covalently agonist-bound states differ in their ability to gate the ion channel. Ultimately, an attractive future direction would be the use of the tethering approach combined with X-ray crystallographic studies to resolve the structure of these covalent agonist-bound states.
Materials and Methods
Chemical Synthesis.
Synthesis of NCS-ATP was adapted from ref. 26 and is detailed in SI Materials and Methods. NCS-Ado was obtained using a similar procedure as for NCS-ATP. All chemical products were characterized by 1H-, 31P-NMR, and UV spectrum.
Complementary DNA Construction and Site-Directed Mutagenesis.
The pcDNA-based expression plasmids, mutagenesis, and sequencing procedure have been described previously (5).
Cell Culture and Transfection.
HEK-293 cells were cultured and transiently transfected with the rat P2X2 constructs (0.01–0.3 μg) and a green fluorescent protein cDNA construct (0.3 μg), as described previously (5).
Cell-Surface Labeling.
Cell-surface expression of L186E mutant was determined as described previously (5) using the thiol-cleavable reagent sulfosuccinimidyl-2-(biotinamido)ethyl-1,3-dithiopropionate (Sulfo-NHS-SS-Biotin, Pierce).
Whole-Cell Recordings.
Patch pipettes (3–5 MΩ) contained (in mM) 140 KCl, 5 MgCl2, 5 EGTA, 10 Hepes, pH 7.3. External solution contained (in mM) 140 NaCl, 2.8 KCl, 2 CaCl2, 2 MgCl2, 10 glucose, 10 Hepes, pH 7.3. The holding potential was -60 mV. Dose-response relationship experiments and drug applications were carried out as described previously (5). The apparent reaction rate constant kapp was determined by fitting data with a single exponential equation. KD and kmodification were determined by fitting data with the following equation:
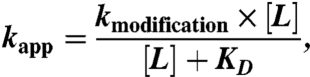 |
[1] |
in which L is the concentration of NCS-ATP, kmodification is the covalent modification rate constant, and KD is the reversible dissociation constant of NCS-ATP.
Single-Channel Recordings.
Single-channel recordings using outside-out configuration were made from HEK-293 cells at room temperature 24–72 h after transfection. Recording pipettes pulled from borosilicate glass (Harvard Apparatus) were coated with Sylgard 184 (Dow Corning Co.) and fire polished to yield resistances of 6–20 MΩ. The holding potential was -120 mV. The extracellular solution contained (in mM) 147 NaCl, 2 KCl, 1 CaCl2, 1 MgCl2, 10 Hepes, and 13 glucose, pH 7.3. The intracellular solution contained (in mM) 147 NaF, 10 Hepes, and 10 EGTA, pH 7.3. The osmolarity was approximately 300 mosmol kg-1 for all the solutions. Following NCS-ATP exposure, patches were always washed by the external solution for at least 15 s before recordings.
Data were acquired with a patch-clamp amplifier (HEKA EPC 10) using PATCHMASTER software (HEKA Co.), sampled at 4–10 kHz, and low-pass filtered at 2.9 kHz. For off-line analysis, data were refiltered to give a cascaded filter cutoff frequency of 1–2 kHz. Channel events were detected by using a 50% threshold criterion (30) and idealized with TAC (Bruxton Co.). Nominal open probability (NPo) was determined with TACFit (Bruxton Co.) and was defined as the fraction of time for which the channels are open, where N is the number of active channels in the patch and Po is the single-channel open probability. As previously observed for WT P2X2 receptor under ATP activation (10), flickery bursts were also visible with ill-defined conductance levels. Such flickering openings were treated as previously specified (10): Each burst was defined as a single conductance state possessing a lot of noise.
Molecular Modeling.
Homology modeling of cysteine mutants was performed with MODELLER 9.7 (31). Docking was performed with the software Autodock vina (32).
Supplementary Material
Acknowledgments.
We thank Prof. M. Goeldner, Prof. S. J. Edelstein, and Dr. B. Winsor for critical reading of the manuscript. This work was supported by the Agence Nationale de la Recherche Grant 06-0050-01 and the Centre National de la Recherche Scientifique (CNRS) (Programme d’Incitation à la Mobilité d’Équipe). R.J. is a recipient of a fellowship from the China Scholarship Council, and S.G. is a recipient of a fellowship from the CNRS.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1102170108/-/DCSupplemental.
References
- 1.Browne LE, Jiang LH, North RA. New structure enlivens interest in P2X receptors. Trends Pharmacol Sci. 2010;31:229–237. doi: 10.1016/j.tips.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Surprenant A, North RA. Signaling at purinergic P2X receptors. Annu Rev Physiol. 2009;71:333–359. doi: 10.1146/annurev.physiol.70.113006.100630. [DOI] [PubMed] [Google Scholar]
- 3.Jiang LH, et al. Subunit arrangement in P2X receptors. J Neurosci. 2003;23:8903–8910. doi: 10.1523/JNEUROSCI.23-26-08903.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagaya N, Tittle RK, Saar N, Dellal SS, Hume RI. An intersubunit zinc binding site in rat P2X2 receptors. J Biol Chem. 2005;280:25982–25993. doi: 10.1074/jbc.M504545200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiang R, et al. A putative extracellular salt bridge at the subunit interface contributes to the ion channel function of the ATP-gated P2X2 receptor. J Biol Chem. 2010;285:15805–15815. doi: 10.1074/jbc.M110.101980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cao L, Broomhead HE, Young MT, North RA. Polar residues in the second transmembrane domain of the rat P2X2 receptor that affect spontaneous gating, unitary conductance, and rectification. J Neurosci. 2009;29:14257–14264. doi: 10.1523/JNEUROSCI.4403-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kracun S, Chaptal V, Abramson J, Khakh BS. Gated access to the pore of a P2X receptor: Structural implications for closed-open transitions. J Biol Chem. 2010;285:10110–10121. doi: 10.1074/jbc.M109.089185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li M, Kawate T, Silberberg SD, Swartz KJ. Pore-opening mechansim in trimeric P2X receptor channels. Nat Commun. 2010;1:1–7. doi: 10.1038/ncomms1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kawate T, Michel JC, Birdsong WT, Gouaux E. Crystal structure of the ATP-gated P2X(4) ion channel in the closed state. Nature. 2009;460:592–598. doi: 10.1038/nature08198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding S, Sachs F. Single channel properties of P2X2 purinoceptors. J Gen Physiol. 1999;113:695–720. doi: 10.1085/jgp.113.5.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Evans RJ. Structural interpretation of P2X receptor mutagenesis studies on drug action. Br J Pharmacol. 2010;161:961–971. doi: 10.1111/j.1476-5381.2010.00728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang LH, Rassendren F, Surprenant A, North RA. Identification of amino acid residues contributing to the ATP-binding site of a purinergic P2X receptor. J Biol Chem. 2000;275:34190–34196. doi: 10.1074/jbc.M005481200. [DOI] [PubMed] [Google Scholar]
- 13.Ennion S, Hagan S, Evans RJ. The role of positively charged amino acids in ATP recognition by human P2X1 receptors. J Biol Chem. 2000;275:29361–29367. doi: 10.1074/jbc.M003637200. [DOI] [PubMed] [Google Scholar]
- 14.Roberts JA, Evans RJ. ATP binding at human P2X1 receptors. Contribution of aromatic and basic amino acids revealed using mutagenesis and partial agonists. J Biol Chem. 2004;279:9043–9055. doi: 10.1074/jbc.M308964200. [DOI] [PubMed] [Google Scholar]
- 15.Wilkinson WJ, Jiang LH, Surprenant A, North RA. Role of ectodomain lysines in the subunits of the heteromeric P2X2/3 receptor. Mol Pharmacol. 2006;70:1159–1163. doi: 10.1124/mol.106.026658. [DOI] [PubMed] [Google Scholar]
- 16.Roberts JA, Evans RJ. Cysteine substitution mutants give structural insight and identify ATP binding and activation sites at P2X receptors. J Neurosci. 2007;27:4072–4082. doi: 10.1523/JNEUROSCI.2310-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marquez-Klaka B, Rettinger J, Bhargava Y, Eisele T, Nicke A. Identification of an intersubunit cross-link between substituted cysteine residues located in the putative ATP binding site of the P2X1 receptor. J Neurosci. 2007;27:1456–1466. doi: 10.1523/JNEUROSCI.3105-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roberts JA, et al. Cysteine substitution mutagenesis and the effects of methanethiosulfonate reagents at P2X2 and P2X4 receptors support a core common mode of ATP action at P2X receptors. J Biol Chem. 2008;283:20126–20136. doi: 10.1074/jbc.M800294200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roberts JA, Valente M, Allsopp RC, Watt D, Evans RJ. Contribution of the region Glu181 to Val200 of the extracellular loop of the human P2X1 receptor to agonist binding and gating revealed using cysteine scanning mutagenesis. J Neurochem. 2009;109:1042–1052. doi: 10.1111/j.1471-4159.2009.06035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foucaud B, Perret P, Grutter T, Goeldner M. Cysteine mutants as chemical sensors for ligand-receptor interactions. Trends Pharmacol Sci. 2001;22:170–173. doi: 10.1016/s0165-6147(00)01674-6. [DOI] [PubMed] [Google Scholar]
- 21.Sullivan DA, Cohen JB. Mapping the agonist binding site of the nicotinic acetylcholine receptor. Orientation requirements for activation by covalent agonist. J Biol Chem. 2000;275:12651–12660. doi: 10.1074/jbc.275.17.12651. [DOI] [PubMed] [Google Scholar]
- 22.Foucaud B, et al. Structural model of the N-methyl-D-aspartate receptor glycine site probed by site-directed chemical coupling. J Biol Chem. 2003;278:24011–24017. doi: 10.1074/jbc.M300219200. [DOI] [PubMed] [Google Scholar]
- 23.Mony L, et al. Structural basis of NR2B-selective antagonist recognition by N-methyl-D-aspartate receptors. Mol Pharmacol. 2009;75:60–74. doi: 10.1124/mol.108.050971. [DOI] [PubMed] [Google Scholar]
- 24.Baur R, et al. Covalent modification of GABAA receptor isoforms by a diazepam analogue provides evidence for a novel benzodiazepine binding site that prevents modulation by these drugs. J Neurochem. 2008;106:2353–2363. doi: 10.1111/j.1471-4159.2008.05574.x. [DOI] [PubMed] [Google Scholar]
- 25.Perret P, et al. Interaction of non-competitive blockers within the gamma-aminobutyric acid type A chloride channel using chemically reactive probes as chemical sensors for cysteine mutants. J Biol Chem. 1999;274:25350–25354. doi: 10.1074/jbc.274.36.25350. [DOI] [PubMed] [Google Scholar]
- 26.Scheiner-Bobis G, Mertens W, Willeke M, Schoner W. Synthesis and biochemical characterization of the new sulfhydryl-reactive ATP analogue 8-thiocyano-ATP. Its interaction with Na,K-ATPase and kinases. Biochemistry. 1992;31:2107–2113. doi: 10.1021/bi00122a031. [DOI] [PubMed] [Google Scholar]
- 27.Bo X, Fischer B, Maillard M, Jacobson KA, Burnstock G. Comparative studies on the affinities of ATP derivatives for P2x-purinoceptors in rat urinary bladder. Br J Pharmacol. 1994;112:1151–1159. doi: 10.1111/j.1476-5381.1994.tb13204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trujillo CA, et al. Inhibition mechanism of the recombinant rat P2X(2) receptor in glial cells by suramin and TNP-ATP. Biochemistry. 2006;45:224–233. doi: 10.1021/bi051517w. [DOI] [PubMed] [Google Scholar]
- 29.Ruiz ML, Karpen JW. Single cyclic nucleotide-gated channels locked in different ligand-bound states. Nature. 1997;389:389–392. doi: 10.1038/38744. [DOI] [PubMed] [Google Scholar]
- 30.Colquhoun D, Sigworth FJ. Fitting and statistical analysis of single-channel records. In: Sakmann B, Neher E, editors. Single-Channel Recording. New York: Plenum; 1995. pp. 483–587. [Google Scholar]
- 31.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 32.Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.