

Abstract
Cannabis is the most commonly used illicit substance among pregnant women. Human epidemiological and animal studies have found that prenatal cannabis exposure influences brain development and can have long-lasting impacts on cognitive functions. Exploration of the therapeutic potential of cannabis-based medicines and synthetic cannabinoid compounds has given us much insight into the physiological roles of endogenous ligands (endocannabinoids) and their receptors. In this article, we examine human longitudinal cohort studies that document the long-term influence of prenatal exposure to cannabis, followed by an overview of the molecular composition of the endocannabinoid system and the temporal and spatial changes in their expression during brain development. How endocannabinoid signaling modulates fundamental developmental processes such as cell proliferation, neurogenesis, migration and axonal pathfinding are also summarized.
Keywords: brain development, cannabinoid receptor, cannabis, cortex, endocannabinoids, THC
Cannabis is the world's third most popular recreational drug, after alcohol and tobacco [201]. The hallmarks of its effects are euphoria and relaxation, perceptual alterations, time distortion, appetite inducement and the intensification of ordinary sensory experiences [1]. Cannabis preparations are largely derived from the female plant of Cannabis sativa, and consist of approximately 60 plant-derived cannabinoid compounds (phytocannabinoids), with Δ9-tetrahydrocannabinol (THC) being the predominant psychoactive constituent [2]. Efforts aimed at understanding how THC produces its psychoactive effects have led to the discovery of the endocannabinoid system [3].
The endocannabinoid system is comprised of endogenous cannabinoids (endocannabinoids [eCBs]), the metabolic enzymes responsible for the formation and degradation of eCBs, and the cannabinoid receptors and their interacting proteins [4,5]. eCB signaling is involved in a myriad of physiological processes including retrograde signaling and modulation of synaptic function in the CNS, and analgesic and metabolic effects on lipid profile and glucose homeostasis in the periphery [6–11]. Indeed, several therapeutic effects have been ascribed to compounds targeting the endocannabinoid system, including treatment of pain, affective and neurodegenerative disorders, gastrointestinal inflammation, obesity and related metabolic dysfunctions, cardiovascular conditions and liver diseases [12,13]. Synthetic THC (dronabinol) is approved in the USA to alleviate the emesis and nausea associated with cancer and chemotherapy, and weight loss associated with HIV infection. Clinical trials are underway to determine whether cannabis-based compounds are effective in the treatment of multiple sclerosis [14] and neuropathic pain [15]. Sativex, a pharmaceutical preparation containing the psychoactive THC with the nonpsychotropic cannabidiol in approximately a 1:1 weight ratio, was approved in Canada for the treatment of neuropathic pain associated with multiple sclerosis, and in England for the spasticity associated with multiple sclerosis [16]. Furthermore, several forms of pharmacological manipulation of the endocannabinoid system, including synthetic cannabinoid receptor agonists and antagonists and inhibitors of endocannabinoid degradation are undergoing clinical development [17–20].
The increasing popularity of cannabis consumption among young people between 15 and 30 years of age, the critical period for adolescent brain development, has raised concerns over the health consequences of cannabis use. In addition, cannabis is the most commonly abused illicit drug in pregnant women in Western societies [202]. Given the lipophilic nature of THC, it is estimated that one-third of THC in the plasma crosses the fetoplacental barrier [21], and is secreted through the breast milk [22]. Given that the THC content of confiscated cannabis samples has increased substantially over the past 20 years [23], fetuses of cannabis-using mothers could be exposed to significant amounts of THC during the perinatal period. Therefore, cannabis abuse is potentially deleterious to the children of cannabis-using mothers through abnormal brain development owing to exogenous cannabis exposure during the perinatal period. This article will focus on the neurobehavioral consequences of prenatal cannabis exposure in humans.
A central role for eCB signaling in brain development is now emerging [7,24,25]. Perinatal and adolescent cannabis exposure may disrupt the precise temporal and spatial control of eCB signaling at critical stages of neural development, leading to detrimental effects on later nervous system functioning. Indeed, longitudinal studies in humans with prenatal cannabis exposure demonstrated exaggerated startle response and poor habituation to novel stimuli in infants, and hyperactivity, inattention and impaired executive function in adolescents [26–29]. Many of these behavioral effects have also been modeled in animal studies [30]. Furthermore, possible teratogenic effects of endocannabinoid system-based therapies in pregnant women and long-term exposure to eCB signaling-modifying agents such as organophosphate pesticides need to be taken into consideration.
This article aims to summarize the existing literature on the behavioral consequences of prenatal exposure to the phytocannabinoid THC, summarizing key findings from epidemiological studies in humans. Experimental studies in rodents have been reviewed extensively elsewhere and will be only briefly discussed here [29–32]. The molecular composition of the endocannabinoid system and their temporal and spatial distributions in embryonic brain in humans and rodents are also summarized. Finally, experimental evidence demonstrating how eCB signaling in this molecular framework affects specific events in developing neural circuits is discussed.
Adverse effect of prenatal exposure to marijuana
Cannabis use during 2010 for those aged 15–64 was estimated to be between 2.9 and 4.3% worldwide, with a high but steady occurrence in North America and Western/Central Europe [203]. The Substance Abuse and Mental Health Services Administration estimates that 7.1% of pregnant women aged 18–25 have used illicit drugs in the month prior to being surveyed [201]. Marijuana was the most prevalent substance abused, ranging from 2–6% usage as determined by interview or self-report [33,34]. However, one study on cannabis usage during pregnancy found an 11% usage rate by measuring serum metabolites [35] close to that seen in age-matched, non-pregnant women (10.9%, [201]). Moore et.al. found that within a British population, marijuana was the only illicit drug pregnant women were likely to continue using to term [36].
Available data linking prenatal cannabis exposure to congenital anomalies or preterm delivery are weak. While fetal alcohol syndrome-like features in prenatally cannabis-exposed newborns have been reported [37], a number of other studies have failed to replicate this finding [38–40]. Nevertheless, prenatal cannabis exposure has been found to be associated with fetal growth restriction [41,42], and learning disabilities and memory impairment in the exposed offspring [43–45]. The mean potency of cannabis preparations, in terms of contents of its psychoactive constituent, THC, has increased from 3.4% in 1993 to 8.8% in 2008, and can reach as high as 30% in certain hashish preparations [23]. This fact is important since THC effects are dose related and classical studies carried out in the 1970s used doses that refected cannabis intake at that period of time. Key findings from human and animal studies regarding behavioral consequences of cannabis exposure during pregnancy and/or lactation will be summarized in the following section.
Human studies
Despite the fact that marijuana is the most widely used illicit drug by pregnant women, there are few studies on the prevalence of prenatal drug exposure. Most information is derived from two longitudinal cohort studies, the Ottawa Prenatal Prospective Study (OPPS) and the Maternal Health Practices and Child Development Study (MHPCD). OPPS, initiated in 1978, focused on assessing prenatal exposure effects of tobacco and marijuana in a low-risk, mainly Caucasian, predominantly middle-class Canadian cohort [46]. Initiated in 1982, the MHPCD focused mainly on prenatal alcohol and marijuana exposure in a group of women from Pittsburgh, Pennsylvania. These women were generally of low socioeconomic status and comprised of approximately half Caucasian and half African–American ethnicity [47]. In both the OPPS and MHPCD studies, cannabis use during pregnancy was not associated with increased miscarriage rates, premature deliveries or any other complications (Table 1). Physically, marijuana exposure was not correlated with any changes in head circumference at the mid-gestational stage (17–22 weeks), although a significant reduction in foot length and bodyweight at this gestational period was reported [48]. These changes in bodyweight and foot length were not present at birth [35], although head circumference was reportedly larger in the exposed cohort at 8 months [49]. These anthropometric measurements were used as an indication of normal fetal development, which correlates with brain development [50].
Table 1.
Prenantal marijuana exposure studies in humans.
| Age† | Cohort‡ | Category | Results/outcomes | Exposure details | Ref. |
|---|---|---|---|---|---|
| Fetus | |||||
| 17-22 GW | – | Growth | Reduced foot length and bodyweight | ≥0.4 J/day | [48] |
| 18-22 GW | – | Dopamine signaling | Decreased D2 mRNA in amygdala of males only | ≥0.4 J/day | [152] |
| 18-22 GW | – | Endorphin signaling | Decreased opioid peptide (PENK) and receptor (κ) in caudal putamen and thalamus, respectively; increased opioid receptor (μ) in the amygdala | – | [74] |
| Neonatal | |||||
| Neonatal | OPPS | Neurobehavior | Increased tremors, exaggerated startles and diminished responsiveness to light | – | [46] |
| Neonata | MHPCD | Growth | Decreased body length | ≥1 J/day, first | [49] |
| Neonata | – | Growth | No effect on birthweight, length or gestational age | Trim | [54] |
| Neonatal | VIPS | Neurobehavior Growth | No effectNo effect on birthweight, preterm delivery or abruptio placentae | – | [35] |
| Neonata | – | Growth | No effect on birthweight, length or gestational age | – | [52] |
| Behavior | More irritable, less responsive to calming, increased jitters and startles | – | |||
| Neonatal | NBDPS | Growth | No effect on birthweight, gestational age or preterm delivery | – | [40] |
| 1 mo | – | Behavior | Less irritable, more alert, more robust autonomic and motor systems, more autonomically stable and increased orientation | ≥2.86 J/day | [54] |
| Toddler | |||||
| 8 mo | MHPCD | Growth | No effect | – | [49] |
| 9 mo | MHPCD | Mental/motor skills | Delayed mental development | ≥1 J/day, third Trim | [26] |
| 1 y | OPPS | Mental/motor skills | No effect | – | [55] |
| 1 y | – | Mental/motor skills | Decreased motor scores, no effect on mental development | ≥0.5 J/day, first Trim | [149] |
| 19 mo | MHPCD | Mental/motor skills | No effect | – | [26] |
| 2 y | OPPS | Mental/motor skills | No effect | – | [55] |
| 3 y | MHPCD | Intelligence | No effect on overall IQ for entire cohort | – | [57] |
| Cognition | For African–Americans: decrease in short-term memory and verbal reasoning | ≥1 J/day, first/second Trim | |||
| 3 y | MHPCD | Sleep and arousal | Lowered sleep efficiency, more nocturnal arousals and more awake time after sleep onset | – | [153] |
| Childhood | |||||
| 4 y | – | Sustained attention | Increased number of omission errors | First Trim | [45] |
| 4 y | MHPCD | Motor skills | No effect on balance and coordination skills | – | [154] |
| 5-6 y | OPPS | Cognition and language | No effect | – | [155] |
| 6 y | MHPCD | Growth | No effect | – | [156] |
| 6 y | OPPS | Memory | No effect | ≥0.86 J/day | [58] |
| Attention | Increased number of omission errors | ||||
| Behavior | Described as more impulsive and hyperactive | ||||
| 6 y | MHPCD | Impulsivity | Increase in errors of ommission | Second Trim | [60] |
| Sustained attention | Decrease in errors of omission | ||||
| 6 y | MHPCD | Intelligence | Lower overall composite score | ≥1 J/day, first/second Trim | [59] |
| Cognition | Lower verbal reasoning, quantitative reasoning and short-term memory | ||||
| 9–12 y | OPPS | Reading and language | No effect in regards to reading or language | – | [68] |
| 9–12 y | OPPS | Intelligence | No effect in terms of full scale IQ | – | [63] |
| Executive function | Impulse control and visual hypothesis aspects are negatively impacted | > 0.86 J/day | |||
| Adolescence | |||||
| 10 y | MHPCD | Behavior and emotion | Increased inattention, hyperactivity, impulsivity, delinquent behavior and externalizing problems | ≥0.4 J/day, | [62] |
| Behavior and emotion | Fewer internalizing problems, although not correlated with teacher's report | first/third Trim ≥0.4 J/day, | |||
| 10 y | MHPCD | Learning and memory | Predicted lower scores in design memory and screening index | second Trim ≥0.89 J/day, | [47] |
| Sustained attention | Increase in errors of commission | first Trim ≥0.89 J/day, | |||
| 10 y | MHPCD | Depression | Increased levels of depressive symptoms | second Trim >0.89 J/day, | [64] |
| 12y | – | Psychotic symptoms | No effect | first/third Trim | [66] |
| 10–14 y | – | Volumetric MRI | No effect on cortical gray matter volume, white matter volume, cerebral spinal fluid or parenchymal volume | – | [69] |
| 10 y | MHPCD | Behavior and cognition | Negatively associated with depressive symptoms, IQ, learning and memory | ≥0.89 J/day, first/second Trim | [61] |
| 14 y | Delinquent behaviors | Increased delinquent behaviors | ≥0.89 J/day | ||
| 13–16 y | OPPS | Sustained attention | Decreased stability of attention over time | ≥0.86 J/day | [71] |
| 13–16 y | OPPS | Growth | No changes in weight, height or puberty symptoms | – | [157] |
| 13–16 y | OPPS | Visual memory | Lower scores in abstract designs and Peabody spelling | ≥0.86 J/day | [27] |
| 16y | MHPCD | Fine motor coordination | Various light deficits in processing speed and interhemispheric motor coordination | >2 J/mo | [158] |
| Visual-motor coordination | Slight increase in visual-motor coordination | ||||
| Young adult | |||||
| 18-22y | OPPS | Response inhibition | Slightly more errors of commission | – | [72] |
| Response inhibition by fMRI | Increased bilateral PFC activity, right premotor cortex activity; decreased activity in left cerebellum | – | |||
| Intelligence | No effect | – | |||
| Working memory by fMRI | Increased activity in left medial PFC, inferior frontal gyrus and left cerebellum; decreased activity in right brain regions - medial PFC, DLPFC and ventrolateral PFC | – | [73] | ||
Exposed offspring study age.
Specified conditions of prenatal exposure.
DLPFC: Dorsolateral prefrontal cortex; GW: Gestation week(s); J: Joint; MHPCD: Maternal Health Practices and Child Development Project; mo: Month(s); NBDPS: National Birth Defects Prevention Study; OPPS: Ottawa Prenatal Prospective Study; PENK: Proenkephalin; PFC: Prefrontal cortex; Trim: Trimester; VIPS: Vaginal Infections and Prematurity Study; y: Year(s).
The OPPS study found that prenatal marijuana exposure was highly correlated with an increase in exaggerated startles and tremors as well as with a significant reduction in habituation to light at the neonatal stage [46,51]. Altered sleep patterns were found in the MHPCD study, and the authors also reported a non-significant trend towards increased irritability [49]. A study on neonates from adolescent mothers found in cannabis-exposed infants transiently increased irritability, excitability and arousal 24–72 h after birth [52]. However, these symptoms were not reported within the MHPCD cohort [53] or in an ethnographic field study based in Jamaica [54]. The MHPCD cohort also demonstrated that a higher amount of cannabis use per day (defined as more than one joint per day) during the third trimester of pregnancy was associated with decreased mental scores of the Bayley Scales of Infant Development at 9 months of age, a difference that disappeared by 18 months [26]. No cognitive deficit was observed during early childhood in the OPPS study, particularly between the ages of 1 and 3 years, suggesting that CNS abnormalities might be absent or subclinical in toddlers [55,56].
For 3–4 year old children, prenatal marijuana exposure negatively affected the verbal and memory domains in both the OPPS and MHPCD studied groups. Cognitive development assessed by the Stanford-Binet Intelligence Scale demonstrated a negative association of short-term memory and verbal reasoning with first and/or second trimester marijuana usage [57]. Similarly, memory and verbal domains, measured by the McCarthy Scales of Children's Abilities, decreased with daily marijuana usage [56]. However, composite intelligence scores in both studies were not impacted at this age by maternal marijuana use.
When children reach school age at around 5–6 years old, reports on the consequences of prenatal marijuana exposure begin to diverge. Exposed children from the OPPS cohort appear to have no memory deficits [58], while those from the MHPCD cohort report short-term memory deficits that correlate strongly with heavy second trimester exposure [59]. Cannabis-exposed children in the OPPS cohort scored significantly lower in tests for sustained attention, while those from the MHPCD group actually displayed increased attention (measured by fewer errors of omission in a continuous performance task) from second trimester exposure [60]. Both groups reported an increase in impulsive and hyperactive behaviors. Follow-up studies found that problems of depression, hyperactivity, inattention and impulsivity persist into the 9–12 year age range [47,61–64], raising speculation of deficits in higher cognitive processes such as executive function [65].
Upon closer inspection, the impact of prenatal marijuana exposure is a little more difficult to discern. For example, one report from the MHPCD cohort found that heavy first- and third-trimester exposure (rated as >0.89 joints/day) was associated with increased hyperactivity and impulsivity [62], while another found that heavy second trimester exposure was significantly associated with increased impulsivity [47]. First- and third-trimester exposure also predicted increased levels of depressive symptoms, assessed by the Children's Depression Inventory [61,64], whereas second-trimester usage was associated with some depressive, but fewer internalizing, symptoms compared with the extent observed in first- and third-trimester exposure groups [62]. Verbal IQ, reading comprehension, overall IQ, presence of psychotic symptoms and sleep patterns do not seem to be impacted [63,66–68]. A recent study has assessed volumetric changes using functional MRI (fMRI) in the brains of children exposed to a number of drugs, including marijuana, during pregnancy. This study found evidence of reduced cortical gray matter and parenchymal volume in children (aged 10- to 14-years old) with intra-uterine marijuana exposure [69].
Executive functions comprise capacities such as cognitive flexibility, sustained and focused attention, and working memory; these can not be assessed with global, standardized tests of cognition [70]. Data from both OPPS and MHPCD cohorts demonstrated deficits in executive functions, which seemed to persist into late adolescence and young adulthood in children of cannabis users [47,63]. The two tests that were found to be negatively affected in marijuana-exposed children both involve the visual analysis and impulse control aspects of executive functions [63]. In the OPPS cohort, 13–16 year olds that were heavily exposed (rated as >0.86 joints/day) displayed deficits in visual memory, visual analysis [27] and the ability to maintain attention (referred to as stability) [71]. fMRI studies with Go/No–Go paradigms conducted to assess response inhibition with 18–22 year-old subjects from the OPPS group found that prenatal exposure was associated with alterations of neural activity in various brain areas during certain tasks [72]. fMRI analysis of visuospatial working memory tasks with the same group also revealed significant changes in levels of activity in the cannabis-exposed group [73]. Peculiarly, prenatal exposure had both positive and negative associations with fMRI response; whereas mostly left brain regions experienced an increase in activity, right brain regions experienced the opposite during tasks. Whether these differences in regional activation/deactivation are a result of various compensatory mechanisms, or if these changes reflect a behavioral alteration that can only be observed with different or more sensitive testing requires further investigation.
Data addressing whether prenatal marijuana exposure can clearly alter the structural and molecular composition of the fetal brain are scarce. Hurd et al. developed a post-mortem human fetal brain collection of midgestational subjects with maternal cannabis use that has begun to provide the first insights into the molecular and biochemical alterations associated with prenatal cannabis exposure on human neuro-development [48]. In the mid gestation human fetus, prenatal cannabis exposure was associated with decreased pro-enkephalin mRNA levels in the striatum, increased μ-opioid receptor expression in the amygdala and reduced κ-opioid receptor mRNA levels in the mediodorsal thalamus [74]. These data suggest that striatal enkephalin/D2 receptor and the opioid system in the limbic-related structures are vulnerable to prenatal cannabis exposure.
In summary, cannabis consumption during pregnancy has profound but variable effects on offspring in several areas of cognitive development [28]. Most of the information on the long-term consequences of prenatal exposure to cannabis comes from longitudinal studies of the OPPS and MHPCD cohorts. By comparing data from the cohorts, a pattern emerges where maternal cannabis use is associated with impaired high-order cognitive function in the offspring, including attention deficits and impaired visuoperceptual integration. It is possible that genetic and environmental interactions may affect the extent of long-term neurobehavioral deficits resulting from prenatal exposure. Recent advances in methodology in prenatal substance use research employ novel approaches to disentangle the exposure to substance effects from correlated risk factors [75]. For example, in the prospective Generation R Study, where 7452 mothers were enrolled during pregnancy and information on substance use and ultrasound measures of fetal growth in early, mid- and late pregnancy were collected, information on paternal cannabis use was also included [41]. Thus, maternal cannabis use during pregnancy was associated with growth restriction in mid and late pregnancy, and also with lower birthweight, while no such association was found for paternal cannabis use in the same period, demonstrating a direct biological effect of maternal intrauterine exposure to cannabis on fetal growth [41]. Refined study designs and novel approaches will assist in confirming and extending the findings of associations between prenatal cannabis exposure and offspring outcomes [75].
Animal studies
Epidemiological studies on long-term neurobehavioral effects of drugs of abuse are subject to a number of confounding factors such as dosage, poly-substance abuse, length and frequency of drug usage, pregnancy stage and environmental factors such as maternal nutrition and socioeconomic problems, commonly associated with drug abuse. Animal models provide tighter experimental control over these factors and a wealth of data have been generated on the behavioral and molecular changes associated with prenatal exposure to cannabis preparations or synthetic compounds (termed ‘cannabinoids’ for the following discussion). Overall, pre- and early post-natal exposure to cannabinoids lead to changes in social interactions, novelty responses and memory in the adult offspring [29–32]. In addition, drug addictive behaviors are modified in cannabinoid-exposed offspring, as indicated by sensitized responses to the reinforcing effects of heroin and morphine in conditioned place preference tests [76,77]. While the exact molecular pathways underlying these behavioral changes are not clear, numerous studies demonstrated that prenatal cannabinoid exposure may lead to alterations in GABAergic, glutamatergic, dopaminergic, serotoninergic and opioidergic systems in offspring [28,32,78]. In addition, perinatal cannabinoid exposure disrupts neurodevelopment through modifications of gene expression [79] involved in neuronal specification and synapse physiology [80].
Endocannabinoid system during neural development
Endocannabinoid signaling plays important roles in learning and memory, anxiety, depression, addiction, appetite and feeding behaviors, pain and neuroprotection [5,13]. In the adult brain, eCBs are synthesized and released ‘on demand’ from postsynaptic neuronal compartments, where they act as retrograde messengers by engaging CB1 cannabinoid receptors (CB1R) on presynaptic terminals [6,8,81] to attenuate neurotransmitter release in many excitatory and inhibitory synapses. Examples of eCB-dependent synaptic plasticity include depolarization-induced suppression of inhibition and excitation, metabotropic suppression of inhibition and excitation, and some forms of long-term depression [8]. However, recent findings establish a strikingly different molecular organization of eCB signaling networks in the developing mammalian forebrain. The following subsections summarize the temporal and spatial distribution of various components of the endocannabinoid system in developing brains with an emphasis on the developmental profile of CB1R as it is responsible for mediating most of the effects of THC [1,82].
Overview of the endocannabinoid system
Endocannabinoids are amides, esters and ethers of long-chain polyunsaturated fatty acids. There are two major families of eCBs, acyl amides and acyl esters. Anandamide (arachidonoyl ethanolamide [AEA]) and 2-arachidonoyl glycerol (2-AG), respectively, are the prototypical members of each family. The enzymes that synthesize AEA are still uncertain, with at least four routes of AEA biosynthesis proposed to occur in brain homogenates [83]. These enzymes include N-acyl-phosphatidyl-ethanolamine-specific phospholipase D, α,β-hydrolase domain-containing 4, glycerophosphodiesterase-1 and phosphatases such as PTPN22 [5,84,85]. These biosynthetic pathways demonstrate substantial overlap and may be able to substitute for one another. AEA is hydrolyzed to arachidonic acid and ethanolamine by fatty acid amide hydrolase (FAAH) [15,86]. 2-AG is synthesized by sn-1-selective diacylglycerol lipases α and β (DAGLα and DAGLβ) [87]. Recent data from genetic ablation of these two isoforms suggest DAGLα is the major CNS form in adult mice and is important for postsynaptic release of 2-AG to transiently suppress GABA-mediated transmission at inhibitory synapses in the hippocampus [88]. Interestingly, DAGLα mRNA level is found to be decreased in the hippocampus of epileptic human patients, while DAGLβ isoform levels were unchanged, suggesting that under pathophysiological conditions, DAGLα is the affected isoform [89]. However, it remains to be determined which DAGL isoform is responsible for 2-AG production in humans. 2-AG is hydrolyzed to arachidonic acid and glycerol by monoglyceride lipase (MAGL) [90] and α/β-hydrolase domain-containing serine hydrolases (ABHD6/12) [91].
The eCBs elicit diverse central and peripheral effects by activating the cannabinoid receptors: CB1R and CB2R[92,93], G-protein coupled receptor 55 (GPR55) [94–97], the transient receptor potential of vanilloid type-1 (TRPV1) channel, the peroxisome proliferators-activated receptor (PPAR)α [4] and at least two, as yet molecularly uncharacterized, receptors [98]. The signal transduction mechanisms include Gi-mediated inhibition of adenylyl cyclase and modulation of ion channels. Cannabinoid signaling often inhibits voltage-dependent Ca2+ channels (N and P/Q type) or activates inwardly rectifying K+ channels [99]. In addition, cannabinoids stimulate various signaling pathways involved in the regulation of cell fate, such as the MAP kinase family (ERK, JNK and p38), protein kinase B and the sphingolipid pathway [100,101].
Ontogeny of the endocannabinoid system
Owing to their lipophilic nature, endocannabinoids are highly unstable and difficult to quantify, hence the paucity of data on endocannabinoid levels during development. The only available data comes from older studies employing mass spectrometry, where levels of AEA and 2-AG have been shown to vary substantially in rodent brains throughout prenatal development [78,102]. AEA is present at low concentrations in the brain at midgestation and gradually increases through the perinatal period until adult levels are reached [102], whereas fetal 2-AG levels gradually increase through the prenatal period, with a surge occurring at birth [102,103]. Notably, 2-AG concentrations (2–8 nmol/g tissue) are approximately 1000 fold higher than those of AEA (3–6 pmol/g tissue) throughout brain development [102]. However, additional studies are required to substantiate these findings.
The ontogeny of the metabolizing enzymes and receptors of the endocannabinoid system has not been extensively characterized. Nevertheless, current data suggest that the endocannabinoid system exists from the earliest stage of pregnancy, in the preimplantation embryo and uterus [104], placenta [105] and in the developing fetal brain [78], presenting multiple points of vulnerability to exogenous cannabis or cannabimimetic drug exposure throughout gestation. In the mouse, stimulation of CB1R arrests the development of two-cell embryos into blastocytsts in culture [104]. AEA is present in the pregnant uterus at relatively high levels (5–10 nmol/g tissue) that fluctuate with changes in the pregnancy status, with higher levels associated with a nonreceptive uterine environment [104]. Indeed, low levels of the AEA-degrading enzyme FAAH and high levels of CB1R expression in human placenta are associated with spontaneous miscarriage [106]. Studies characterizing the endocannabinoid system in early human pregnancy (weeks 7–12 gestation) demon strated that CB2R and FAAH are expressed in relatively constant levels in trophoblasts in early gestation, but their cellular distribution changed from syncytio trophoblast to the mesenchymal core of the villus [105].
In the developing mouse brain, CB1R are expressed as early as day 11 postgestation (comparable with 5–6-week old human embryos), with gradually increasing levels of both mRNA and receptor density (revealed by radiolabeled agonist binding) throughout the prenatal period in the whole brain [78] . CB1R are abundantly expressed in corticolimbic areas of the fetal rodent brain [78]. Pharmacological studies of the ability of the CB1R agonist, WIN55212-2, to stimulate [35S] GTPγS binding indicate that CB1Rs are functionally active from early stages of development [107]. In human fetal brains, CB1Rs were detected at week 14 of gestation, with preferential expression in the cerebral cortex, hippocampus, caudate nucleus, putamen and cerebellar cortex. By week 20, intense expression is evident in CA2–CA3 of hippocampus and in the basal nuclear group of the amygdala [107,108].
More recently, several specific antibodies against different endocannabinoid system components have become available, making it possible to examine the distribution of specific enzymes at the light- and electron-microscopic level. In particular, immunohistological studies using specific antibodies have mapped the temporal and spatial distribution of CB1R, DAGLα/β and MAGL during neural development in mice [103,109,110]. Similar to ligand binding and mRNA expression studies, CB1R immunoreactivity can be detected by embryonic day (E)12.5, and is localized to reelin-expressing Cajal-Retzius cells and newly differentiated postmitotic glutamatergic neurons of the mouse telencephalon [111], and to the subpial area of the ganglionic eminence and marginal zone of the neocortex [112]. From E13.5 to birth, abundant CB1R immunoreactivity is detected in several long-range axonal tracts including corticofugal tracts such as corticothalamic (Figure 1) and corticospinal tracts [109,111]. On the subcellular level, CB1R is localized to somato-dendritic endosomes at E12.5 and then to developing axons of glutamatergic neurons at E13.5 and after this time[111] . The ‘atypical’ pattern of CB1R expression in long-range glutamatergic axons disappears after birth [110,111]. During late gestation (E17–18), CB1R immunoreactivity becomes detectable in axons and axonal growth cones of cholecystokinin (CCK)-positive GABAergic interneurons [113,114]. The origin of these CB1R containing interneurons has been traced to the caudal ganglionic eminence and pallial–subpallial boundary at E11–12. These cells undergo a complex long-distance migration, first radially to the marginal zone, then tangentially in the lateral-to-medial direction within the dorsal telencephalon, eventually reaching their final destination in the cortex, hippocampus and dentate gyrus where they migrate radially and differentiate into CB1/CCK+ or CB1/reelin/calretinin+ GABAergic interneurons [112].
Figure 1. Expression pattern of CB1R, DAGLβ and MAGL in developing thalamocortical axonal tracts.
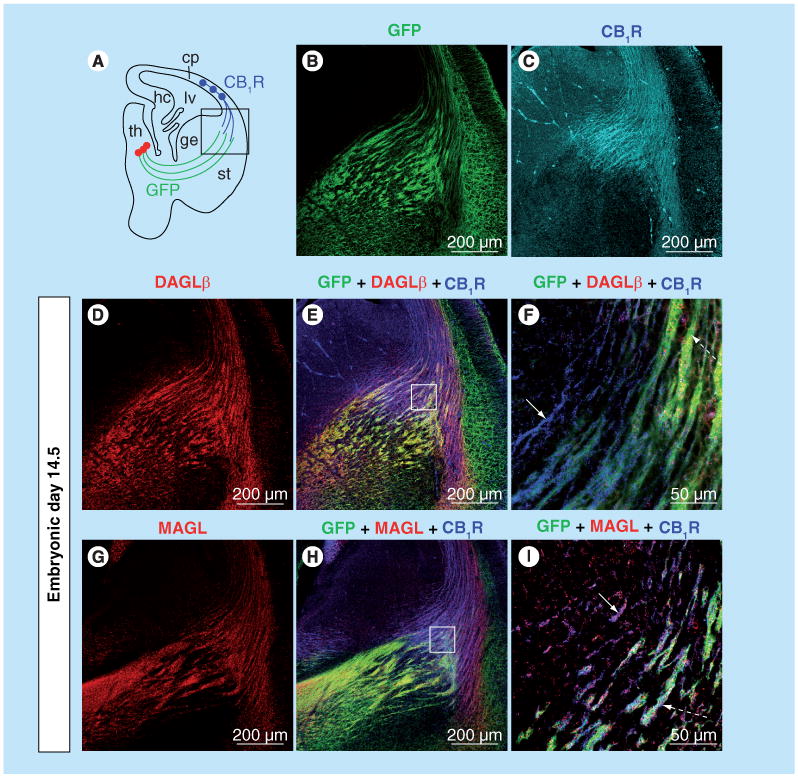
(A) Embryonic brain slice highlighting the path of developing thalamocortical axons (green lines) and corticothalamic axons (blue lines) at E14.5. A thalamocortical axon reporter mice line (TCAmGFP) was generated by crossing a Cre-reporter line containing a foxed ‘stop transcription’ sequence in front of membrane-anchored green fuorescent protein (mGFP) followed by an IRES-NLS-lacZ gene inserted into exon 2 of the Tau locus with RORα-Cre mice. (B) Thalamocortical axons extending toward the cortex are GFP labeled in TCAmGFP reporter mice. (C) Using the TCAmGFP mice, CB1R is demonstrated to be localized to corticothalamic, but not thalamocortical, axons during brain development. (D–I) DAGLβ is localized mainly to GFP-labeled thalamocortical axons (dashed arrow in [F]), while MAGL is localized to both CB1R-containing corticothalamic (arrow in I) and GFP-labeled thalamocortical axons (dashed arrow in [I]). (F & I) higher magnifcation of squared areas in (E & H). CB1R: CB1 cannabinoid receptor; cp: Cortical plate; DAGLβ: Diacylglycerol lipase β; ge: Ganglionic eminence; GFP: Green fuorescent protein; hc: Hippocampus; lv: Lateral ventricle; MAGL: Monoglyceride lipase; RORα: Retinoic acid-receptor-related orphan receptor α; st: Striatum; TCA: Thalamocortical axon; th: Thalamus.
The overall protein levels of CB1Rs are relatively constant throughout forebrain development, while DAGLα protein levels peak at E14.5/E16.5 and then dramatically decrease in neonates, furthermore MAGL transiently decreases around E18.5 (Figure 2) [103]. Similar to CB1R, the distribution of DAGLα/β and MAGL are localized to long-range glutamatergic axons in the prenatal period (Figure 1) [103,110]. Interestingly, after E16.5, MAGL expression undergoes a dramatic change from cortical plate and long-range axon tracts to a restricted expression in perisomatic segments and proximal dendrites both in the late-gestational brain and at birth [103]. This coincides with the surge in cortical and hippocampal 2-AG concentrations, suggesting that MAGL plays an essential role in determining 2-AG availability in the developing brain.
Figure 2. Temporal changes in the expression of endocannabinoid system components during development.

DAGLα and MAGL are expressed starting at midgestation. Expression levels of DAGLα decrease from E18.5 onwards, while there is a transient decrease in MAGL expression levels around birth. CB1R is expressed at relatively constant levels throughout brain development and, prenatally, is primarily localized to the corticofugal tract. The expression pattern undergoes a dramatic change after birth, with the adult-like distribution pattern in the cortex is apparent after P1. CB1R: CB1 cannabinoid receptor; DAGLα: Diacylglycerol lipase α; MAGL: Monoglyceride lipase; P1: Postnatal day 1.
Together, these expression studies indicate that components of the endocannabinoid system are expressed early in life and are positioned to modulate neuronal generation, differentiation, migration and neural circuit wiring during development.
Involvement of endocannabinoid signaling in neural development
During embryogenesis, the cerebral cortex develops from the rostral part of the neural tube designated the telencephalic pallium. In rodents, pyramidal neurons originate from the cortical ventricular zone (VZ) whereas interneurons are generated in the ganglionic eminence of the basal telencephalon [115,116]. Neurons generated early in the VZ migrate radially towards the surface of the cerebral vesicles to form the primordial plexiform layer or preplate (Figure 3). Neurons generated later migrate to form a layer within the preplate, the so-called cortical plate (CP), thus splitting it into a superficial marginal zone (layer I) and a deep subplate. The neurons of the CP assemble into layers II–VI in an ‘inside-out’ sequence: the deepest cellular layers are assembled frst and those closest to the surface last (Figure 3). The nonpyramidal cells originate predominantly in the medial ganglionic eminence and migrate tangentially following parallel migratory streams, in the subventricular zone (SVZ), intermediate zone and marginal zone, and progressively enter the developing CP. Accumulating evidence indicates that eCBs regulate several aspects of neural development, including neurogenesis, neuronal migration, neurite outgrowth and axonal pathfinding. Much of the insights came from studies in genetic ablation of CB1R and FAAH (increasing the endogenous levels of AEA) in mice and from pharmacological manipulations (CB1R blockade and inhibition of FAAH). Recent findings on the involvement of AEA- and 2-AG-mediated signaling through CB1R in various aspects of neural development will be summarized in the following section.
Figure 3. Generation of neuronal diversity in cortical formation.

The earliest born neurons form the PP, which is later split into the superfcial MZ and the deeper layer SP. Neuroprogenitors residing in the VZ and SVZ in mice produce pyramidal projection neurons in an ‘inside-out’ fashion, migrating radially towards the surface to populate the cp. The cp develops inbetween the MZ and SP and gives rise to the multilayered pattern, while the progenitor zones progressively reduce in size. The relationship between different populations of cells is depicted at E16.5. Radial glial cells divide symmetrically in the VZ to produce additional radial glial cells. A fraction of these depart from the VZ and migrate radially towards the pial surface, giving rise to different types of projection neurons. Interneurons migrate tangentially from the ganglionic eminence and enter the neocortex around E17. cp: Cortical plate; ge: Ganglionic eminence; hc: Hippocampus; lv: Lateral ventricle; MZ: Marginal zone; PP: Preplate; SP: subplate; st: Striatum; SVZ: Subventricular zone; th: Thalamus; VZ: Ventricular zone.
Cell proliferation, neurogenesis & oligodendrogliogenesis
During mammalian embryogenesis, the generation of the CNS relies on a finely regulated balance of neuroprogenitor proliferation, differentiation and survival that is controlled by a number of extracellular signaling cues [117]. In the adult brain, identification of neuroprogenitor cells in the subgranular zone supports the existence of hippocampal neurogenesis, which is implicated in several brain functions including learning and memory, depression and brain repair [118–120].
Several lines of evidence support a role for eCBs in neural progenitor proliferation [24,121–123]. The expression of CB1R, FAAH, DAGL and MAGL in VZ/SVZ neuroprogenitor cells has been reported [103,109,121,122,124] . CB1R activation promotes progenitor cell proliferation, while genetic deletion of CB1R decreases cortical progenitor proliferation in VZ/SVZ in the embryonic brain [121]. By contrast, deletion of FAAH increases neural progenitor proliferation in the embryonic brain [121] and inhibition of FAAH by the inhibitor URB597 increased VZ/SVZ progenitor proliferation in embryonic brain slices [109]. Thus, the reciprocal consequences of blockading CB1R and increasing AEA suggest that local AEA levels acting through CB1R, modulate neural progenitor proliferation in the embryonic brain.
The effect of CB1R activation on neurogenesis has been examined in early postnatal and adult brains and in neurospheres, with conflicting results (reviewed in [24,125]). It should be noted that the net effect of cannabinoids on neurogenesis during postnatal development, in adults and in culture, is probably influenced by the status of nervous system maturation, where inherent characteristics of the neural progenitors may differ. Increased proliferation of neural progenitors is observed in the hippocampus of adult FAAH knockout mice [121], consistent with the observation of decreased neuroprogenitor proliferation in the hippocampus of both early postnatal and adult CB1R knockout mice [122]. Furthermore, using a kainate-induced excitotoxicity model, excitotoxicity-induced hippocampal neural progenitor proliferation is abrogated in CB1R knockout mice and in wildtype mice treated with the CB1R antagonist, SR141716 [126]. HU210, a synthetic CB1R agonist, increases adult hippocampal neurogenesis and exerts anxiolytic and antide-pressant effects in rats [127], while the synthetic CB1R/CB2R agonist, WIN55212-2, partially restores hippocampal neurogenesis in the aged rat brain [128]. By contrast, methanandamide, a non-hydrolyzable AEA analog, significantly decreases neurogenesis in the adult hippocampus of rats [129]. Moreover, AEA decreases the expression of a mature neuronal marker and inhibits neurite outgrowth of cortical neural progenitors in vitro, while the CB1R antagonist SR141716 increases the rates of neuronal differentiation of neural progenitors [129]. The same group later reported that activation of CB1R on neural progenitors promotes the differentiation of the latter into glia cells [122]. Together, these studies suggest an endogenous AEA tone that actively modulates neural progenitor differentiation through the CB1R. Furthermore, a role for 2-AG in adult neurogenesis has been demonstrated [88,124]. A DAGL antagonist inhibits the proliferation of cultured neural stem cells, and the proliferation of progenitor cells in young adult mice, and adult neurogenesis in the SVZ and hippocampus, is impaired in both DAGLα and DAGLβ knockout mice [88,124]. Interestingly, a sex difference in cell proliferation in developing rat amygdala, mediated by 2-AG, has recently been reported [130]. Newborn females had higher rates of cell proliferation than males, which were abrogated by inhibition of MAGL in females [130]. The impact of prenatal THC exposure on neurogenesis remains to be examined.
Cannabinoid signaling has also been suggested to participate in postnatal myelination processes [131–133]. Postnatal myelination involves radial migration of astrocyte-like (type B) precursor cells from the SVZ to the overlying white matter, where these cells are differentiated into astrocytes and oligodendrocytes (reviewed in [134,135]). CB1R is expressed in radial glia-like cells and B-like type cells, while CB2R is expressed in a subpopulation of SVZ cells containing the polysialylated neural cell adhesion mole cule [122,132]. The 2-AG synthesizing enzymes, DAGLα and β, and degrading enzyme, MAGL, are also found in oligodendrocytes in various differentiation stages[136]. Agonist stimulation of CB1R and CB2R increases the expression of myelin basic protein in subcortical white matter [132]. Furthermore, cannabinoid signaling has been suggested to participate in adult oligodendrogenesis after toxic or autoimmune demyelinating lesions, when precursor cells are recruited from the SVZ towards the injured area and give rise to oligodendrocytes [14,131,133].
Neuronal migration
Following mitosis, newborn pyramidal progenitor cells in the VZ/SVZ migrate radially into the cortical plate and populate distinct cortical layers [116]. Proper regulation of this migration is important for appropriate cortical patterning. Genetic deletion or blockade of CB1R leads to delayed migration of cortical neurons. In cultured brain slices, elevating AEA levels through FAAH gene removal or pharmacological blockade enhances the migration of newly borne postmitotic neurons into the cortical plate [109]. In contrast to pyramidal neurons, cortical inhibitory interneurons migrate tangentially from the ganglionic eminences to the cortical plate. CB1R signaling is also implicated in the regulation of the long-distance migration CCK-positive interneurons [112,113]. Stimulation with AEA and WIN55,212–2, in cooperation with brain-derived neurotrophic factor, a major prodifferentiating neurotrophin for this cell class, induces the long-distance migration of GABA-containing interneurons in the ganglionic eminence [137]. Prenatal THC has been demonstrated to increase the density of CCK-expressing interneurons in the rat hippocampus in vivo [137]. Thus, eCBs modulate both migration of cortical principal neurons and certain classes of interneurons. Increases in local AEA concentration probably affect the proper placement of pyramidal neurons and/or CCK+ basket cells. Furthermore, DAGLα and MAGL have been demonstrated to be expressed in mouse migratory neuroblasts that travel along the rostral migratory stream to populate the olfactory bulb, and DAGL inhibition results in decreased migration in scratch wound assays and in explant cultures, suggesting a role for 2-AG in regulating cell migration following adult neurogenesis [138].
Axon pathfinding & fasciculation
Once pyramidal neurons reach their final destination, they must project their axons to connect to their postsynaptic partners. Axon tracts navigate along stereotyped pathways, and fasciculate and defasciculate in distinctive domains along their path [139,140]. The formation of precise neural circuits requires orchestrated interactions between axon tracts, and between the navigating axonal growth cones and the environmental cues at distinctive locations. Genetic deletion of CB1R or prenatal CB1R pharmacological blockade in mice led to increases in the number of axons with aberrant trajectories in the corpus callosum and to abnormal fasciculation of long-range axons [109,110]. Similarly, knockdown of CB1R in zebrafish leads to abnormal axonal fasciculation [141].
Thalamic axons projecting into the cortex provide the majority of cortical sensory input, while reciprocal innervations from the cortex to the thalamus send critical feed-back to modulate the thalamic responses required to perform the complex information gathering and integration that underlie sensory processing [142–144]. A ‘handshake hypothesis’, which proposes that thalamocortical axons and corticothalamic axons interact and serve as scaffolds to guide each other to their final destinations, has been postulated [145–147]. Recent data suggest that the endocannabinoid system may be modulating this handshake interaction [110]. When thalamic and cortical axons meet and intermingle in the basal telencephalon during development, CB1R is localized to corticothalamic axons, while the 2-AG synthesizing enzymes, DAGLα/β and degrading enzyme MAGL, are present in both thalamocortical and corticothalamic axons (Figure 1) [103,110]. Thus, 2-AG could be produced in both axonal tracts to act in both an autocrine and paracrine fashion, while MAGL may serve to restrict 2-AG availability. Interestingly, genetic deletion of CB1R in cortical neurons leads to aberrant fasciculation in both corticothalamic and thalamocortical axons despite normal target recognition [110], suggesting that 2-AG-mediated signaling at CB1R may be modulating the fasciculation process during the handshake interaction between cortical and thalamic axons.
More recently, a role for CB1R in axonal guidance has been demonstrated in the retinal system [148]. Eye-specific segregation of retinal projections in the dorsal lateral geniculate nucleus of thalamus is impaired in CB1R knockout mice. Furthermore, CB1R appears to act in concert with the adhesion molecule deleted in colorectal cancer (DCC; a receptor for axonal guidance molecule, netrin-1) to influence axonal growth cone behavior [148].
At the subcellular level, a recent morphometric study provided evidence for a microgradient of 2-AG in elongating axons [106]. While MAGL is coexpressed with both CB1R and DAGLα in cultured cortical neurons, MAGL is differentially recruited to distinct subcellular domains, particularly in the consolidated axon shaft. In this paradigm, CB1Rs are maintained in a state of inactivity by the absence of 2-AG (owing to presence of MAGL) while undergoing vesicular transport along the consolidated axon. The absence of MAGL at the growth cones lifts the restriction on CB1R signaling and promotes cell-autonomous axonal growth. This scheme may serve to prevent ectopic branching and axon guidance errors, since in vitro study in pyramidal cells found that CB1R activation leads to increased neurite branching [112].
These findings firmly demonstrate multiple roles for the endocannabinoid system in brain development. A detailed knowledge of eCB signaling is important in understanding the long-term consequences of alterations in CB1R activity during neurodevelopment, a potential etiology for the mental health disorders linked to prenatal or adolescent cannabis use, or following therapeutic manipulations of the endocannabinoid system.
Conclusion
Marijuana abuse during pregnancy and adolescence represents a major health problem owing to its potential consequences on neural development. Prenatally cannabis-exposed children display cognitive deficits, suggesting that maternal consumption has interfered with the proper maturation of the brain. Several pharmacological effects of THC, the active principle of Cannabis sativa preparations such as hashish and marijuana, are mimicked by the endogenous eCBs, 2-AG and AEA. On the other hand, pharmacological inhibition or genetic deletion of either FAAH or MAGL to elevate endogenous levels of AEA and 2-AG, respectively, does not reproduce the full spectrum of responses observed with THC. Interestingly, a recently developed dual FAAH/MAGL inhibitor (JZL195) induced THC-like drug discrimination responses, which were not observed with disruption of either FAAH or MAGL alone [149]. The observation that the THC-like additive effects of dual FAAH/MAGL blockade can be reversed by CB1R antagonists suggests that most, if not all, acute cognitive responses to THC are mediated through the CB1R [150] . Whether THC produces effect by partially activating CB1R or antagonizing the action of 2-AG and AEA at this receptor remains an open question [151]
2-AG is the most abundant eCB in the brain. Basal 2-AG levels in the adult brain are approximately 200 times those of AEA [83], while an approximately 1000-fold excess of tissue 2-AG concentrations over those of AEA exists in the fetal brain [102]. The relative contribution of the two arms of eCB signaling in regulating neurogenesis, axonal growth and guidance, and synaptogenesis during development is not well understood. The data from dual blockade of FAAH/MAGL indicate that AEA and 2-AG signaling pathways interact to regulate specific behavioral processes in vivo, including those relevant to drug abuse [150]. Hence, future studies employing novel pharmacological and genetic tools may help to dissect out specific eCB signaling pathways in regulating neural developmental processes.
Future perspective
Data from human epidemiological and animal studies during prenatal exposure to cannabis, together with experimental data studying the physiological roles of endocannabinoid signaling, point to the importance of this system in modulating and fine-tuning brain development. Future challenges include detailed mapping of the expression profiles of various endocannabinoid components, utilizing newly available genetic knockout mice and target-specific pharmacological reagents, elucidations of pathway/s involved in each aspect of neural developmental processes. Furthermore, the relative contribution of anandamide and 2-AG in activation of CB1R signaling, and how exogenous phytocannabinoids such as THC interfere in this signaling network remain open questions. A better understanding of the molecular framework of endocannabinoid signaling may contribute to defning the molecular changes underlying the neurobehavioral changes observed in the offspring of cannabis users and the neurodevelopmental impact of the therapeutic manipulation of the endocannabinoid system during pregnancy.
Medscape: Continuing Medical Education Online.
This activity has been planned and implemented in accordance with the Essential Areas and policies of the Accreditation Council for Continuing Medical Education through the joint sponsorship of Medscape, LLC and Future Medicine Ltd. Medscape, LLC is accredited by the ACCME to provide continuing medical education for physicians.
Medscape, LLC designates this Journal-based CME activity for a maximum of 1 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
All other clinicians completing this activity will be issued a certificate of participation. To participate in this journal CME activity: (1) review the learning objectives and author disclosures; (2) study the education content; (3) take the post-test with a 70% minimum passing score and complete the evaluation at www.medscape.org/journal/fnl; (4) view/print certificate.
Learning objectives.
Upon completion of this activity, participants should be able to:
Describe the epidemiology of prenatal exposure to marijuana, based on a review
Describe the neurodevelopmental effects of prenatal exposure to marijuana
Describe the effects of endocannabinoids on neural development and how prenatal exposure to marijuana influences these effects
Executive Summary.
Adverse effect of prenatal exposure to marijuana
Marijuana is the most prevalent illicit substance abused by pregnant women, with an incidence of 2–6% (determined by interview or self-report) and as high as 11 % by serum.
The mean potency of marijuana preparations, in terms of Δ9-tetrahydrocannabinol content, has increased from 3.4% in 1993 to 8.8% in 2008, reaching 30% in some hashish preparations.
Human marijuana consumption during pregnancy appears to have lasting effects on the child's higher cognitive function.
Endocannabinoid system during neural development
The endocannabinoid system is comprised of endogenous cannabinoids (endocannabinoids), the metabolic enzymes responsible for their formation and degradation, and the cannabinoid receptors and their interacting proteins.
Anandamide and 2-arachidonoyl glycerol (2-AG) are the best-studied endocannabinoids. Levels of these two endocannabinoids increases gradually during development, with 2-AG concentrations approximately 1000-fold higher than anandamide.
CB1 cannabinoid receptors (CB1Rs) are expressed in the cerebral cortex, hippocampus, caudate nucleus, putamen and cerebellar cortex in the human fetal brain.
CB1R is highly expressed in navigating corticofugal axons during development.
Involvement of endocannabinoid signaling in neural development
CB1R activation promotes neural progenitor cell proliferation.
Anandamide may modulate neural progenitor differentiation.
CB1R activation promotes radial migration of pyramidal neurons.
CB1R signaling modulates the fasciculation of long-range axon bundles.
CB1R participates in the ‘handshake’ between developing corticothalamic and thalamocortical axons.
Acknowledgments
Authors and Credentials: Chia-Shan Wu, PhD, The Cain Foundation Laboratories, Jan and Dan Duncan Neurological Research Institute, Texas Children's Hospital. Disclosure: Chia-Shan Wu, PhD, has disclosed no relevant financial relationships. Christopher P Jew, The Cain Foundation Laboratories, Jan and Dan Duncan Neurological Research Institute, Texas Children's Hospital; Program in Developmental Biology, Baylor College of Medicine, Houston, Texas. Disclosure: Christopher P Jew has disclosed no relevant financial relationships. Hui-Chen Lu, PhD, The Cain Foundation Laboratories, Jan and Dan Duncan Neurological Research Institute, Texas Children's Hospital; Department of Pediatrics; Program in Developmental Biology; Department of Neuroscience, Baylor College of Medicine, Houston, Texas. Disclosure: This work was supported by NIH grants: NS048884 (NINDS), DA029381 (NIDA) and HD065561 (NICHD) to H-C Lu.
Footnotes
Financial & competing interests disclosure: Editor: Elisa Manzotti, Editorial Director, Future Science Group. Disclosure: Elisa Manzotti has disclosed no relevant financial relationships.
CME author: Laurie Barclay, Freelance writer and reviewer, Medscape, LLC. Disclosure: Laurie Barclay, MD, has disclosed no relevant financial relationships.
No writing assistance was utilized in the production of this manuscript.
Bibliography
Papers of special note have been highlighted as:
■ of interest
■■ of considerable interest
- 1.Hall W, Solowij N. Adverse effects of cannabis. Lancet. 1998;352(9140):1611–1616. doi: 10.1016/S0140-6736(98)05021-1. [DOI] [PubMed] [Google Scholar]
- 2.Adams IB, Martin BR. Cannabis: pharmacology and toxicology in animals and humans. Addiction. 1996;91(11):1585–1614. [PubMed] [Google Scholar]
- 3.Mechoulam R, Fride E, Di Marzo V. Endocannabinoids. Eur J Pharmacol. 1998;359(1):1–18. doi: 10.1016/s0014-2999(98)00649-9. [DOI] [PubMed] [Google Scholar]
- 4■■.Mackie K, Stella N. Cannabinoid receptors and endocannabinoids: evidence for new players. AAPS J. 2006;8(2):E298–E306. doi: 10.1007/BF02854900. Overview of the endocannabinoid system and evidence for new cannabinoid receptors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Piomelli D. The molecular logic of endocannabinoid signalling. Nat Rev Neurosci. 2003;4(11):873–884. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- 6.Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev. 2003;83(3):1017–1066. doi: 10.1152/physrev.00004.2003. [DOI] [PubMed] [Google Scholar]
- 7■■.Harkany T, Guzman M, Galve-Roperh I, Berghuis P, Devi LA, Mackie K. The emerging functions of endocannabinoid signaling during CNS development. Trends Pharmacol Sci. 2007;28(2):83–92. doi: 10.1016/j.tips.2006.12.004. Overview of the endocannabinoid pathways controlling neuronal specifcation during brain development. [DOI] [PubMed] [Google Scholar]
- 8.Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev. 2009;89(1):309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- 9.Katona I, Freund TF. Endocannabinoid signaling as a synaptic circuit breaker in neurological disease. Nat Med. 2008;14(9):923–930. doi: 10.1038/nm.f.1869. [DOI] [PubMed] [Google Scholar]
- 10.Osei-Hyiaman D, Harvey-White J, Batkai S, Kunos G. The role of the endocannabinoid system in the control of energy homeostasis. Int J Obes (Lond) 2006;30(Suppl. 1):S33–S38. doi: 10.1038/sj.ijo.0803276. [DOI] [PubMed] [Google Scholar]
- 11.Tam J, Vemuri VK, Liu J, et al. Peripheral CB1 cannabinoid receptor blockade improves cardiometabolic risk in mouse models of obesity. J Clin Invest. 2010;120(8):2953–2966. doi: 10.1172/JCI42551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Di Marzo V. Targeting the endocannabinoid system: to enhance or reduce? Nat Rev Drug Discov. 2008;7(5):438–455. doi: 10.1038/nrd2553. [DOI] [PubMed] [Google Scholar]
- 13.Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58(3):389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arevalo-Martin A, Vela JM, Molina-Holgado E, Borrell J, Guaza C. Therapeutic action of cannabinoids in a murine model of multiple sclerosis. J Neurosci. 2003;23(7):2511–2516. doi: 10.1523/JNEUROSCI.23-07-02511.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cravatt BF, Lichtman AH. Fatty acid amide hydrolase: an emerging therapeutic target in the endocannabinoid system. Curr Opin Chem Biol. 2003;7(4):469–475. doi: 10.1016/s1367-5931(03)00079-6. [DOI] [PubMed] [Google Scholar]
- 16.Barnes MP. Sativex: clinical effcacy and tolerability in the treatment of symptoms of multiple sclerosis and neuropathic pain. Expert Opin Pharmacother. 2006;7(5):607–615. doi: 10.1517/14656566.7.5.607. [DOI] [PubMed] [Google Scholar]
- 17.Janero DR, Makriyannis A. Cannabinoid receptor antagonists: pharmacological opportunities, clinical experience, and translational prognosis. Expert Opin Emerg Drugs. 2009;14(1):43–65. doi: 10.1517/14728210902736568. [DOI] [PubMed] [Google Scholar]
- 18.Maida V, Ennis M, Irani S, Corbo M, Dolzhykov M. Adjunctive nabilone in cancer pain and symptom management: a prospective observational study using propensity scoring. J Support Oncol. 2008;6(3):119–124. [PubMed] [Google Scholar]
- 19.Zutt M, Hanssle H, Emmert S, Neumann C, Kretschmer L. Dronabinol for supportive therapy in patients with malignant melanoma and liver metastases. Hautarzt. 2006;57(5):423–427. doi: 10.1007/s00105-005-1063-x. [DOI] [PubMed] [Google Scholar]
- 20.Ahn K, Johnson DS, Cravatt BF. Fatty acid amide hydrolase as a potential therapeutic target for the treatment of pain and CNS disorders. Expert Opin Drug Discov. 2009;4(7):763–784. doi: 10.1517/17460440903018857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hutchings DE, Martin BR, Gamagaris Z, Miller N, Fico T. Plasma concentrations of Δ-9-tetrahydrocannabinol in dams and fetuses following acute or multiple prenatal dosing in rats. Life Sci. 1989;44(11):697–701. doi: 10.1016/0024-3205(89)90380-9. [DOI] [PubMed] [Google Scholar]
- 22.Perez-Reyes M, Wall ME. Presence of Δ9-tetrahydrocannabinol in human milk. N Engl J Med. 1982;307(13):819–820. doi: 10.1056/NEJM198209233071311. [DOI] [PubMed] [Google Scholar]
- 23.Mehmedic Z, Chandra S, Slade D, et al. Potency trends of Δ9-THC and other cannabinoids in confscated cannabis preparations from 1993 to 2008. J Forensic Sci. 2010;55(5):1209–1217. doi: 10.1111/j.1556-4029.2010.01441.x. [DOI] [PubMed] [Google Scholar]
- 24.Galve-Roperh I, Palazuelos J, Aguado T, Guzman M. The endocannabinoid system and the regulation of neural development: potential implications in psychiatric disorders. Eur Arch Psychiatry Clin Neurosci. 2009;259(7):371–382. doi: 10.1007/s00406-009-0028-y. [DOI] [PubMed] [Google Scholar]
- 25.Harkany T, Keimpema E, Barabas K, Mulder J. Endocannabinoid functions controlling neuronal specifcation during brain development. Mol Cell Endocrinol. 2008;286(1–2 Suppl. 1):S84–S90. doi: 10.1016/j.mce.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 26.Richardson GA, Day NL, Goldschmidt L. Prenatal alcohol, marijuana, and tobacco use: infant mental and motor development. Neurotoxicol Teratol. 1995;17(4):479–487. doi: 10.1016/0892-0362(95)00006-d. [DOI] [PubMed] [Google Scholar]
- 27.Fried PA, Watkinson B, Gray R. Differential effects on cognitive functioning in 13- to 16-year-olds prenatally exposed to cigarettes and marihuana. Neurotoxicol Teratol. 2003;25(4):427–436. doi: 10.1016/s0892-0362(03)00029-1. [DOI] [PubMed] [Google Scholar]
- 28■■.Jutras-Aswad D, Dinieri JA, Harkany T, Hurd YL. Neurobiological consequences of maternal cannabis on human fetal development and its neuropsychiatric outcome. Eur Arch Psychiatry Clin Neurosci. 2009;259(7):395–412. doi: 10.1007/s00406-009-0027-z. A comprehensive review on prenatal marijuana exposure, impacted neurotransmitter systems and neuropsychiatric outcomes. [DOI] [PubMed] [Google Scholar]
- 29■■.Schneider M. Cannabis use in pregnancy and early life and its consequences: animal models. Eur Arch Psychiatry Clin Neurosci. 2009;259(7):383–393. doi: 10.1007/s00406-009-0026-0. Recent and detailed review of animal models of perinatal cannabis exposure, including natural cannabis preparations and Δ-9-tetrahydrocannabinol and synthetic cannabinoid agonists. [DOI] [PubMed] [Google Scholar]
- 30.Navarro M, Rubio P, De Fonseca FR. Behavioural consequences of maternal exposure to natural cannabinoids in rats. Psychopharmacology (Berl) 1995;122(1):1–14. doi: 10.1007/BF02246436. [DOI] [PubMed] [Google Scholar]
- 31■■.Campolongo P, Trezza V, Palmery M, Trabace L, Cuomo V. Developmental exposure to cannabinoids causes subtle and enduring neurofunctional alterations. Int Rev Neurobiol. 2009;85:117–133. doi: 10.1016/S0074-7742(09)85009-5. Recent and detailed review on animal studies summarizing behavioral alterations exhibited by offspring of mothers exposed to cannabis. [DOI] [PubMed] [Google Scholar]
- 32.Trezza V, Cuomo V, Vanderschuren LJ. Cannabis and the developing brain: insights from behavior. Eur J Pharmacol. 2008;585(2–3):441–452. doi: 10.1016/j.ejphar.2008.01.058. [DOI] [PubMed] [Google Scholar]
- 33.Yonkers KA, Gotman N, Kershaw T, Forray A, Howell HB, Rounsaville BJ. Screening for prenatal substance use: development of the Substance Use Risk Profle-Pregnancy scale. Obstet Gynecol. 2010;116(4):827–833. doi: 10.1097/AOG.0b013e3181ed8290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ebrahim SH, Gfroerer J. Pregnancy-related substance use in the United States during 1996–1998. Obstet Gynecol. 2003;101(2):374–379. [Google Scholar]
- 35.Shiono PH, Klebanoff MA, Nugent RP, et al. The impact of cocaine and marijuana use on low birth weight and preterm birth: a multicenter study. Am J Obstet Gynecol. 1995;172(1 Pt 1):19–27. doi: 10.1016/0002-9378(95)90078-0. [DOI] [PubMed] [Google Scholar]
- 36.Moore DG, Turner JD, Parrott AC, et al. During pregnancy, recreational drug-using women stop taking ecstasy (3,4-methylenedioxy-N-methylamphetamine) and reduce alcohol consumption, but continue to smoke tobacco and cannabis: initial fndings from the Development and Infancy Study. J Psychopharmacol. 2010;24(9):1403–1410. doi: 10.1177/0269881109348165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hingson R, Alpert JJ, Day N, et al. Effects of maternal drinking and marijuana use on fetal growth and development. Pediatrics. 1982;70(4):539–546. [PubMed] [Google Scholar]
- 38.Linn S, Schoenbaum SC, Monson RR, Rosner R, Stubblefeld PC, Ryan KJ. The association of marijuana use with outcome of pregnancy. Am J Public Health. 1983;73(10):1161–1164. doi: 10.2105/ajph.73.10.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Gelder MM, Reefhuis J, Caton AR, Werler MM, Druschel CM, Roeleveld N. Maternal periconceptional illicit drug use and the risk of congenital malformations. Epidemiology. 2009;20(1):60–66. doi: 10.1097/EDE.0b013e31818e5930. [DOI] [PubMed] [Google Scholar]
- 40.Van Gelder MM, Reefhuis J, Caton AR, Werler MM, Druschel CM, Roeleveld N. Characteristics of pregnant illicit drug users and associations between cannabis use and perinatal outcome in a population-based study. Drug Alcohol Depend. 2010;109(1–3):243–247. doi: 10.1016/j.drugalcdep.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 41.El Marroun H, Tiemeier H, Steegers EA, et al. Intrauterine Cannabis Exposure Affects Fetal Growth Trajectories: The Generation R Study. J Am Acad Child Adolesc Psychiatry. 2009 doi: 10.1097/CHI.0b013e3181bfa8ee. [DOI] [PubMed] [Google Scholar]
- 42.Zuckerman B, Frank DA, Hingson R, et al. Effects of maternal marijuana and cocaine use on fetal growth. N Engl J Med. 1989;320(12):762–768. doi: 10.1056/NEJM198903233201203. [DOI] [PubMed] [Google Scholar]
- 43.Fried PA, Watkinson B, Gray R. Neurocognitive consequences of marihuana – a comparison with pre-drug performance. Neurotoxicol Teratol. 2005;27(2):231–239. doi: 10.1016/j.ntt.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 44.Goldschmidt L, Richardson GA, Cornelius MD, Day NL. Prenatal marijuana and alcohol exposure and academic achievement at age 10. Neurotoxicol Teratol. 2004;26(4):521–532. doi: 10.1016/j.ntt.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 45.Noland JS, Singer LT, Short EJ, et al. Prenatal drug exposure and selective attention in preschoolers. Neurotoxicol Teratol. 2005;27(3):429–438. doi: 10.1016/j.ntt.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 46.Fried PA, Makin JE. Neonatal behavioural correlates of prenatal exposure to marihuana, cigarettes and alcohol in a low risk population. Neurotoxicol Teratol. 1987;9(1):1–7. doi: 10.1016/0892-0362(87)90062-6. [DOI] [PubMed] [Google Scholar]
- 47.Richardson GA, Ryan C, Willford J, Day NL, Goldschmidt L. Prenatal alcohol and marijuana exposure: effects on neuropsychological outcomes at 10 years. Neurotoxicol Teratol. 2002;24(3):309–320. doi: 10.1016/s0892-0362(02)00193-9. [DOI] [PubMed] [Google Scholar]
- 48.Hurd YL, Wang X, Anderson V, Beck O, Minkoff H, Dow-Edwards D. Marijuana impairs growth in mid-gestation fetuses. Neurotoxicol Teratol. 2005;27(2):221–229. doi: 10.1016/j.ntt.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 49.Day N, Cornelius M, Goldschmidt L, Richardson G, Robles N, Taylor P. The effects of prenatal tobacco and marijuana use on offspring growth from birth through 3 years of age. Neurotoxicol Teratol. 1992;14(6):407–414. doi: 10.1016/0892-0362(92)90051-b. [DOI] [PubMed] [Google Scholar]
- 50.Bendersky M, Alessandri S, Gilbert P, Lewis M. Characteristics of pregnant substance abusers in two cities in the northeast. Am J Drug Alcohol Abuse. 1996;22(3):349–362. doi: 10.3109/00952999609001664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fried PA, Watkinson B, Dillon RF, Dulberg CS. Neonatal neurological status in a low-risk population after prenatal exposure to cigarettes, marijuana, and alcohol. J Dev Behav Pediatr. 1987;8(6):318–326. [PubMed] [Google Scholar]
- 52.De Moraes Barros MC, Guinsburg R, De Araujo Peres C, Mitsuhiro S, Chalem E, Laranjeira RR. Exposure to marijuana during pregnancy alters neurobehavior in the early neonatal period. J Pediatr. 2006;149(6):781–787. doi: 10.1016/j.jpeds.2006.08.046. [DOI] [PubMed] [Google Scholar]
- 53.Richardson GA, Day N, Taylor P. The effect of prenatal alcohol, marijuana, and tobacco exposure on neonatal behavior. Infant Behav Dev. 1989;12:199–209. [Google Scholar]
- 54.Dreher MC, Nugent K, Hudgins R. Prenatal marijuana exposure and neonatal outcomes in Jamaica: an ethnographic study. Pediatrics. 1994;93(2):254–260. [PubMed] [Google Scholar]
- 55.Fried PA, Watkinson B. 12- and 24-month neurobehavioural follow-up of children prenatally exposed to marihuana, cigarettes and alcohol. Neurotoxicol Teratol. 1988;10(4):305–313. doi: 10.1016/0892-0362(88)90032-3. [DOI] [PubMed] [Google Scholar]
- 56.Fried PA, Watkinson B. 36- and 48-month neurobehavioral follow-up of children prenatally exposed to marijuana, cigarettes, and alcohol. J Dev Behav Pediatr. 1990;11(2):49–58. [PubMed] [Google Scholar]
- 57.Day NL, Richardson GA, Goldschmidt L, et al. Effect of prenatal marijuana exposure on the cognitive development of offspring at age three. Neurotoxicol Teratol. 1994;16(2):169–175. doi: 10.1016/0892-0362(94)90114-7. [DOI] [PubMed] [Google Scholar]
- 58.Fried PA, Watkinson B, Gray R. A follow-up study of attentional behavior in 6-year-old children exposed prenatally to marihuana, cigarettes, and alcohol. Neurotoxicol Teratol. 1992;14(5):299–311. doi: 10.1016/0892-0362(92)90036-a. [DOI] [PubMed] [Google Scholar]
- 59.Goldschmidt L, Richardson GA, Willford J, Day NL. Prenatal marijuana exposure and intelligence test performance at age 6. J Am Acad Child Adolesc Psychiatry. 2008;47(3):254–263. doi: 10.1097/CHI.0b013e318160b3f0. [DOI] [PubMed] [Google Scholar]
- 60.Leech SL, Richardson GA, Goldschmidt L, Day NL. Prenatal substance exposure: effects on attention and impulsivity of 6-year-olds. Neurotoxicol Teratol. 1999;21(2):109–118. doi: 10.1016/s0892-0362(98)00042-7. [DOI] [PubMed] [Google Scholar]
- 61■.Day NL, Leech SL, Goldschmidt L. The effects of prenatal marijuana exposure on delinquent behaviors are mediated by measures of neurocognitive functioning. Neurotoxicol Teratol. 2011;33(1):129–136. doi: 10.1016/j.ntt.2010.07.006. Study from the Maternal Health Practices and Child Development Study (MHPCD) cohort showed that adolescents with heavy prenatal marijuana exposure (PME) are almost twice as likely to exhibit delinquent behaviors compared with nonexposed teens. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goldschmidt L, Day NL, Richardson GA. Effects of prenatal marijuana exposure on child behavior problems at age 10. Neurotoxicol Teratol. 2000;22(3):325–336. doi: 10.1016/s0892-0362(00)00066-0. [DOI] [PubMed] [Google Scholar]
- 63.Fried PA, Watkinson B, Gray R. Differential effects on cognitive functioning in 9- to 12-year olds prenatally exposed to cigarettes and marihuana. Neurotoxicol Teratol. 1998;20(3):293–306. doi: 10.1016/s0892-0362(97)00091-3. [DOI] [PubMed] [Google Scholar]
- 64.Gray KA, Day NL, Leech S, Richardson GA. Prenatal marijuana exposure: effect on child depressive symptoms at ten years of age. Neurotoxicol Teratol. 2005;27(3):439–448. doi: 10.1016/j.ntt.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 65.Fried PA. Conceptual issues in behavioral teratology and their application in determining long-term sequelae of prenatal marihuana exposure. J Child Psychol Psychiatry. 2002;43(1):81–102. doi: 10.1111/1469-7610.00005. [DOI] [PubMed] [Google Scholar]
- 66.Zammit S, Thomas K, Thompson A, et al. Maternal tobacco, cannabis and alcohol use during pregnancy and risk of adolescent psychotic symptoms in offspring. Br J Psychiatry. 2009;195(4):294–300. doi: 10.1192/bjp.bp.108.062471. [DOI] [PubMed] [Google Scholar]
- 67.Stone KC, Lagasse LL, Lester BM, et al. Sleep problems in children with prenatal substance exposure: the Maternal Lifestyle study. Arch Pediatr Adolesc Med. 2010;164(5):452–456. doi: 10.1001/archpediatrics.2010.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fried PA, Watkinson B, Siegel LS. Reading and language in 9- to 12-year olds prenatally exposed to cigarettes and marijuana. Neurotoxicol Teratol. 1997;19(3):171–183. doi: 10.1016/s0892-0362(97)00015-9. [DOI] [PubMed] [Google Scholar]
- 69.Rivkin MJ, Davis PE, Lemaster JL, et al. Volumetric MRI study of brain in children with intrauterine exposure to cocaine, alcohol, tobacco, and marijuana. Pediatrics. 2008;121(4):741–750. doi: 10.1542/peds.2007-1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fried PA, Smith AM. A literature review of the consequences of prenatal marihuana exposure An emerging theme of a deficiency in aspects of executive function. Neurotoxicol Teratol. 2001;23(1):1–11. doi: 10.1016/s0892-0362(00)00119-7. [DOI] [PubMed] [Google Scholar]
- 71.Fried PA, Watkinson B. Differential effects on facets of attention in adolescents prenatally exposed to cigarettes and marihuana. Neurotoxicol Teratol. 2001;23(5):421–430. doi: 10.1016/s0892-0362(01)00160-x. [DOI] [PubMed] [Google Scholar]
- 72■■.Smith AM, Fried PA, Hogan MJ, Cameron I. Effects of prenatal marijuana on response inhibition: an fMRI study of young adults. Neurotoxicol Teratol. 2004;26(4):533–542. doi: 10.1016/j.ntt.2004.04.004. The frst functional MRI study on PME individuals demonstrating that altered neuronal activity is retained well into young adulthood. [DOI] [PubMed] [Google Scholar]
- 73.Smith AM, Fried PA, Hogan MJ, Cameron I. Effects of prenatal marijuana on visuospatial working memory: an fMRI study in young adults. Neurotoxicol Teratol. 2006;28(2):286–295. doi: 10.1016/j.ntt.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 74■■.Wang X, Dow-Edwards D, Anderson V, Minkoff H, Hurd YL. Discrete opioid gene expression impairment in the human fetal brain associated with maternal marijuana use. Pharmacogenomics J. 2006;6(4):255–264. doi: 10.1038/sj.tpj.6500375. Along with [152] it is one of the only two human studies to date that describe changes in neurotransmitter signaling by as early as midgestation in response to PME. [DOI] [PubMed] [Google Scholar]
- 75.Huizink AC. Moderate use of alcohol, tobacco and cannabis during pregnancy: new approaches and update on research findings. Reprod Toxicol. 2009;28(2):143–151. doi: 10.1016/j.reprotox.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 76.Spano MS, Ellgren M, Wang X, Hurd YL. Prenatal cannabis exposure increases heroin seeking with allostatic changes in limbic enkephalin systems in adulthood. Biol Psychiatry. 2007;61(4):554–563. doi: 10.1016/j.biopsych.2006.03.073. [DOI] [PubMed] [Google Scholar]
- 77.Spano MS, Fadda P, Fratta W, Fattore L. Cannabinoid-opioid interactions in drug discrimination and self-administration: effect of maternal, postnatal, adolescent and adult exposure to the drugs. Curr Drug Targets. 2010;11(4):450–461. doi: 10.2174/138945010790980295. [DOI] [PubMed] [Google Scholar]
- 78.Fernandez-Ruiz J, Berrendero F, Hernandez ML, Ramos JA. The endogenous cannabinoid system and brain development. Trends Neurosci. 2000;23(1):14–20. doi: 10.1016/s0166-2236(99)01491-5. [DOI] [PubMed] [Google Scholar]
- 79.Colombo G, Rusconi F, Rubino T, et al. Transcriptomic and proteomic analyses of mouse cerebellum reveals alterations in RasGRF1 expression following in vivo chronic treatment with Δ 9-tetrahydrocannabinol. J Mol Neurosci. 2009;37(2):111–122. doi: 10.1007/s12031-008-9114-2. [DOI] [PubMed] [Google Scholar]
- 80.Castaldo P, Magi S, Cataldi M, et al. Altered regulation of glutamate release and decreased functional activity and expression of GLT1 and GLAST glutamate transporters in the hippocampus of adolescent rats perinatally exposed to Δ(9)-THC. Pharmacol Res. 2010;61(4):334–341. doi: 10.1016/j.phrs.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 81.Katona I, Urban GM, Wallace M, et al. Molecular composition of the endocannabinoid system at glutamatergic synapses. J Neurosci. 2006;26(21):5628–5637. doi: 10.1523/JNEUROSCI.0309-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Huestis MA, Gorelick DA, Heishman SJ, et al. Blockade of effects of smoked marijuana by the CB1-selective cannabinoid receptor antagonist SR141716. Arch Gen Psychiatry. 2001;58(4):322–328. doi: 10.1001/archpsyc.58.4.322. [DOI] [PubMed] [Google Scholar]
- 83.Di Marzo V. Endocannabinoid signaling in the brain: biosynthetic mechanisms in the limelight. Nat Neurosci. 2011;14(1):9–15. doi: 10.1038/nn.2720. [DOI] [PubMed] [Google Scholar]
- 84.Liu J, Wang L, Harvey-White J, et al. A biosynthetic pathway for anandamide. Proc Natl Acad Sci USA. 2006;103(36):13345–13350. doi: 10.1073/pnas.0601832103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Simon GM, Cravatt BF. Endocannabinoid biosynthesis proceeding through glycerophospho-N-acyl ethanolamine and a role for α/β-hydrolase 4 in this pathway. J Biol Chem. 2006;281(36):26465–26472. doi: 10.1074/jbc.M604660200. [DOI] [PubMed] [Google Scholar]
- 86.Cravatt BF, Lichtman AH. The enzymatic inactivation of the fatty acid amide class of signaling lipids. Chem Phys Lipids. 2002;121(1–2):135–148. doi: 10.1016/s0009-3084(02)00147-0. [DOI] [PubMed] [Google Scholar]
- 87.Bisogno T, Howell F, Williams G, et al. Cloning of the frst sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J Cell Biol. 2003;163(3):463–468. doi: 10.1083/jcb.200305129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gao Y, Vasilyev DV, Goncalves MB, et al. Loss of retrograde endocannabinoid signaling and reduced adult neurogenesis in diacylglycerol lipase knock-out mice. J Neurosci. 2010;30(6):2017–2024. doi: 10.1523/JNEUROSCI.5693-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ludanyi A, Eross L, Czirjak S, et al. Downregulation of the CB1 cannabinoid receptor and related molecular elements of the endocannabinoid system in epileptic human hippocampus. J Neurosci. 2008;28(12):2976–2990. doi: 10.1523/JNEUROSCI.4465-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dinh TP, Carpenter D, Leslie FM, et al. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci USA. 2002;99(16):10819–10824. doi: 10.1073/pnas.152334899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Marrs WR, Blankman JL, Horne EA, et al. The serine hydrolase ABHD6 controls the accumulation and effcacy of 2-AG at cannabinoid receptors. Nat Neurosci. 2010;13(8):951–957. doi: 10.1038/nn.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346(6284):561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 93.Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365(6441):61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 94.Baker D, Pryce G, Davies WL, Hiley CR. In silico patent searching reveals a new cannabinoid receptor. Trends Pharmacol Sci. 2006;27(1):1–4. doi: 10.1016/j.tips.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 95.Lauckner JE, Jensen JB, Chen HY, Lu HC, Hille B, Mackie K. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc Natl Acad Sci USA. 2008;105(7):2699–2704. doi: 10.1073/pnas.0711278105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pertwee RG. GPR55: a new member of the cannabinoid receptor clan? Br J Pharmacol. 2007;152(7):984–986. doi: 10.1038/sj.bjp.0707464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ryberg E, Larsson N, Sjogren S, et al. The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol. 2007;152(7):1092–1101. doi: 10.1038/sj.bjp.0707460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kreitzer FR, Stella N. The therapeutic potential of novel cannabinoid receptors. Pharmacol Ther. 2009;122(2):83–96. doi: 10.1016/j.pharmthera.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99■■.Mackie K. Signaling via CNS cannabinoid receptors. Mol Cell Endocrinol. 2008;286(1–2 Suppl 1):S60–65. doi: 10.1016/j.mce.2008.01.022. Overview of the signal transduction pathways downstream of cannabinoid receptors in the CNS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Galve-Roperh I, Sanchez C, Cortes ML, Gomez Del Pulgar T, Izquierdo M, Guzman M. Anti-tumoral action of cannabinoids: involvement of sustained ceramide accumulation and extracellular signal-regulated kinase activation. Nat Med. 2000;6(3):313–319. doi: 10.1038/73171. [DOI] [PubMed] [Google Scholar]
- 101.Guzman M, Galve-Roperh I, Sanchez C. Ceramide: a new second messenger of cannabinoid action. Trends Pharmacol Sci. 2001;22(1):19–22. doi: 10.1016/s0165-6147(00)01586-8. [DOI] [PubMed] [Google Scholar]
- 102.Berrendero F, Sepe N, Ramos JA, Di Marzo V, Fernandez-Ruiz JJ. Analysis of cannabinoid receptor binding and mRNA expression and endogenous cannabinoid contents in the developing rat brain during late gestation and early postnatal period. Synapse. 1999;33(3):181–191. doi: 10.1002/(SICI)1098-2396(19990901)33:3<181::AID-SYN3>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 103■.Keimpema E, Barabas K, Morozov YM, et al. Differential subcellular recruitment of monoacylglycerol lipase generates spatial specifcity of 2-arachidonoyl glycerol signaling during axonal pathfnding. J Neurosci. 2010;30(42):13992–14007. doi: 10.1523/JNEUROSCI.2126-10.2010. Article that described temporal and spatial changes in monoglyceride lipase expression and in vitro evidence of a 2-arachidonoyl glycerol microgradient. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Paria BC, Dey SK. Ligand-receptor signaling with endocannabinoids in preimplantation embryo development and implantation. Chem Phys Lipids. 2000;108(1–2):211–220. doi: 10.1016/s0009-3084(00)00197-3. [DOI] [PubMed] [Google Scholar]
- 105.Habayeb OM, Taylor AH, Bell SC, Taylor DJ, Konje JC. Expression of the endocannabinoid system in human frst trimester placenta and its role in trophoblast proliferation. Endocrinology. 2008;149(10):5052–5060. doi: 10.1210/en.2007-1799. [DOI] [PubMed] [Google Scholar]
- 106.Trabucco E, Acone G, Marenna A, et al. Endocannabinoid system in frst trimester placenta: low FAAH and high CB1 expression characterize spontaneous miscarriage. Placenta. 2009;30(6):516–522. doi: 10.1016/j.placenta.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 107.Mato S, Del Olmo E, Pazos A. Ontogenetic development of cannabinoid receptor expression and signal transduction functionality in the human brain. Eur J Neurosci. 2003;17(9):1747–1754. doi: 10.1046/j.1460-9568.2003.02599.x. [DOI] [PubMed] [Google Scholar]
- 108.Wang X, Dow-Edwards D, Keller E, Hurd YL. Preferential limbic expression of the cannabinoid receptor mRNA in the human fetal brain. Neuroscience. 2003;118(3):681–694. doi: 10.1016/s0306-4522(03)00020-4. [DOI] [PubMed] [Google Scholar]
- 109.Mulder J, Aguado T, Keimpema E, et al. Endocannabinoid signaling controls pyramidal cell specifcation and long-range axon patterning. Proc Natl Acad Sci USA. 2008;105(25):8760–8765. doi: 10.1073/pnas.0803545105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110■.Wu CS, Zhu J, Wager-Miller J, et al. Requirement of cannabinoid CB(1) receptors in cortical pyramidal neurons for appropriate development of corticothalamic and thalamocortical projections. Eur J Neurosci. 2010;32(5):693–706. doi: 10.1111/j.1460-9568.2010.07337.x. Article that provided evidence of the involvement of CB1 cannabinoid receptor signaling in the developmental sensory neural circuit and interactions of corticothalamic and thalamocortical projections. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Vitalis T, Laine J, Simon A, Roland A, Leterrier C, Lenkei Z. The type 1 cannabinoid receptor is highly expressed in embryonic cortical projection neurons and negatively regulates neurite growth in vitro. Eur J Neurosci. 2008;28(9):1705–1718. doi: 10.1111/j.1460-9568.2008.06484.x. [DOI] [PubMed] [Google Scholar]
- 112.Morozov YM, Torii M, Rakic P. Origin, early commitment, migratory routes, and destination of cannabinoid type 1 receptor-containing interneurons. Cereb Cortex. 2009;19(Suppl. 1):I78–I89. doi: 10.1093/cercor/bhp028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Berghuis P, Rajnicek AM, Morozov YM, et al. Hardwiring the brain: endocannabinoids shape neuronal connectivity. Science. 2007;316(5828):1212–1216. doi: 10.1126/science.1137406. [DOI] [PubMed] [Google Scholar]
- 114.Morozov YM, Freund TF. Post-natal development of type 1 cannabinoid receptor immunoreactivity in the rat hippocampus. Eur J Neurosci. 2003;18(5):1213–1222. doi: 10.1046/j.1460-9568.2003.02852.x. [DOI] [PubMed] [Google Scholar]
- 115.Marin O, Rubenstein JL. A long, remarkable journey: tangential migration in the telencephalon. Nat Rev Neurosci. 2001;2(11):780–790. doi: 10.1038/35097509. [DOI] [PubMed] [Google Scholar]
- 116.Molyneaux BJ, Arlotta P, Menezes JR, Macklis JD. Neuronal subtype specifcation in the cerebral cortex. Nat Rev Neurosci. 2007;8(6):427–437. doi: 10.1038/nrn2151. [DOI] [PubMed] [Google Scholar]
- 117.Shen Q, Wang Y, Dimos JT, et al. The timing of cortical neurogenesis is encoded within lineages of individual progenitor cells. Nat Neurosci. 2006;9(6):743–751. doi: 10.1038/nn1694. [DOI] [PubMed] [Google Scholar]
- 118.Aimone JB, Deng W, Gage FH. Adult neurogenesis: integrating theories and separating functions. Trends Cogn Sci. 2010;14(7):325–337. doi: 10.1016/j.tics.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Deng W, Aimone JB, Gage FH. New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat Rev Neurosci. 2010;11(5):339–350. doi: 10.1038/nrn2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mu Y, Lee SW, Gage FH. Signaling in adult neurogenesis. Curr Opin Neurobiol. 2010;20(4):416–423. doi: 10.1016/j.conb.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Aguado T, Monory K, Palazuelos J, et al. The endocannabinoid system drives neural progenitor proliferation. FASEB J. 2005;19(12):1704–1706. doi: 10.1096/fj.05-3995fje. [DOI] [PubMed] [Google Scholar]
- 122.Aguado T, Palazuelos J, Monory K, et al. The endocannabinoid system promotes astroglial differentiation by acting on neural progenitor cells. J Neurosci. 2006;26(5):1551–1561. doi: 10.1523/JNEUROSCI.3101-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Guzman M, Sanchez C, Galve-Roperh I. Cannabinoids and cell fate. Pharmacol Ther. 2002;95(2):175–184. doi: 10.1016/s0163-7258(02)00256-5. [DOI] [PubMed] [Google Scholar]
- 124.Goncalves MB, Suetterlin P, Yip P, et al. A diacylglycerol lipase-CB2 cannabinoid pathway regulates adult subventricular zone neurogenesis in an age-dependent manner. Mol Cell Neurosci. 2008;38(4):526–536. doi: 10.1016/j.mcn.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 125.Galve-Roperh I, Aguado T, Palazuelos J, Guzman M. The endocannabinoid system and neurogenesis in health and disease. Neuroscientist. 2007;13(2):109–114. doi: 10.1177/1073858406296407. [DOI] [PubMed] [Google Scholar]
- 126.Aguado T, Romero E, Monory K, et al. The CB1 cannabinoid receptor mediates excitotoxicity-induced neural progenitor proliferation and neurogenesis. J Biol Chem. 2007;282(33):23892–23898. doi: 10.1074/jbc.M700678200. [DOI] [PubMed] [Google Scholar]
- 127.Jiang W, Zhang Y, Xiao L, et al. Cannabinoids promote embryonic and adult hippocampus neurogenesis and produce anxiolytic- and antidepressant-like effects. J Clin Invest. 2005;115(11):3104–3116. doi: 10.1172/JCI25509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Marchalant Y, Brothers HM, Wenk GL. Cannabinoid agonist WIN-55,212–212 partially restores neurogenesis in the aged rat brain. Mol Psychiatry. 2009;14(12):1068–1069. doi: 10.1038/mp.2009.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Rueda D, Navarro B, Martinez-Serrano A, Guzman M, Galve-Roperh I. The endocannabinoid anandamide inhibits neuronal progenitor cell differentiation through attenuation of the Rap1/B-Raf/ERK pathway. J Biol Chem. 2002;277(48):46645–46650. doi: 10.1074/jbc.M206590200. [DOI] [PubMed] [Google Scholar]
- 130.Krebs-Kraft DL, Hill MN, Hillard CJ, McCarthy MM. Sex difference in cell proliferation in developing rat amygdala mediated by endocannabinoids has implications for social behavior. Proc Natl Acad Sci USA. 2010;107(47):20535–20540. doi: 10.1073/pnas.1005003107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Arevalo-Martin A, Garcia-Ovejero D, Molina-Holgado E. The endocannabinoid 2-arachidonoylglycerol reduces lesion expansion and white matter damage after spinal cord injury. Neurobiol Dis. 2010;38(2):304–312. doi: 10.1016/j.nbd.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 132.Arevalo-Martin A, Garcia-Ovejero D, Rubio-Araiz A, Gomez O, Molina-Holgado F, Molina-Holgado E. Cannabinoids modulate Olig2 and polysialylated neural cell adhesion molecule expression in the subventricular zone of post-natal rats through cannabinoid receptor 1 and cannabinoid receptor 2. Eur J Neurosci. 2007;26(6):1548–1559. doi: 10.1111/j.1460-9568.2007.05782.x. [DOI] [PubMed] [Google Scholar]
- 133.Solbrig MV, Fan Y, Hermanowicz N, Morgese MG, Giuffrida A. A synthetic cannabinoid agonist promotes oligodendrogliogenesis during viral encephalitis in rats. Exp Neurol. 2010;226(1):231–241. doi: 10.1016/j.expneurol.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cayre M, Canoll P, Goldman JE. Cell migration in the normal and pathological postnatal mammalian brain. Prog Neurobiol. 2009;88(1):41–63. doi: 10.1016/j.pneurobio.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Goldman JE. Regulation of oligodendrocyte differentiation. Trends Neurosci. 1992;15(10):359–362. doi: 10.1016/0166-2236(92)90179-c. [DOI] [PubMed] [Google Scholar]
- 136.Gomez O, Arevalo-Martin A, Garcia-Ovejero D, et al. The constitutive production of the endocannabinoid 2-arachidonoylglycerol participates in oligodendrocyte differentiation. Glia. 2010;58(16):1913–1927. doi: 10.1002/glia.21061. [DOI] [PubMed] [Google Scholar]
- 137.Berghuis P, Dobszay MB, Wang X, et al. Endocannabinoids regulate interneuron migration and morphogenesis by transactivating the TrkB receptor. Proc Natl Acad Sci USA. 2005;102(52):19115–19120. doi: 10.1073/pnas.0509494102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Oudin MJ, Gajendra S, Williams G, Hobbs C, Lalli G, Doherty P. Endocannabinoids regulate the migration of subventricular zone-derived neuroblasts in the postnatal brain. J Neurosci. 2011;31(11):4000–4011. doi: 10.1523/JNEUROSCI.5483-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Dodd J, Jessell TM. Axon guidance and the patterning of neuronal projections in vertebrates. Science. 1988;242(4879):692–699. doi: 10.1126/science.3055291. [DOI] [PubMed] [Google Scholar]
- 140.Van Vactor D. Adhesion and signaling in axonal fasciculation. Curr Opin Neurobiol. 1998;8(1):80–86. doi: 10.1016/s0959-4388(98)80011-1. [DOI] [PubMed] [Google Scholar]
- 141.Watson S, Chambers D, Hobbs C, Doherty P, Graham A. The endocannabinoid receptor, CB1, is required for normal axonal growth and fasciculation. Mol Cell Neurosci. 2008;38(1):89–97. doi: 10.1016/j.mcn.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 142.Jones EG. Thalamic circuitry and thalamocortical synchrony. Philos Trans R Soc Lond B Biol Sci. 2002;357(1428):1659–1673. doi: 10.1098/rstb.2002.1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Temereanca S, Simons DJ. Functional topography of corticothalamic feedback enhances thalamic spatial response tuning in the somatosensory whisker/barrel system. Neuron. 2004;41(4):639–651. doi: 10.1016/s0896-6273(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 144.Theyel BB, Llano DA, Sherman SM. The corticothalamocortical circuit drives higher-order cortex in the mouse. Nat Neurosci. 2010;13(1):84–88. doi: 10.1038/nn.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hevner RF, Miyashita-Lin E, Rubenstein JL. Cortical and thalamic axon pathfnding defects in Tbr1, Gbt×, and Pa× mutant mice: evidence that cortical and thalamic axons interact and guide each other. J Comp Neurol. 2002;447(1):8–17. doi: 10.1002/cne.10219. [DOI] [PubMed] [Google Scholar]
- 146.Molnar Z, Adams R, Blakemore C. Mechanisms underlying the early establishment of thalamocortical connections in the rat. J Neurosci. 1998;18(15):5723–5745. doi: 10.1523/JNEUROSCI.18-15-05723.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Molnar Z, Blakemore C. How do thalamic axons fnd their way to the cortex? Trends Neurosci. 1995;18(9):389–397. doi: 10.1016/0166-2236(95)93935-q. [DOI] [PubMed] [Google Scholar]
- 148.Argaw A, Duff G, Zabouri N, et al. Concerted action of CB1 cannabinoid receptor and deleted in colorectal cancer in axon guidance. J Neurosci. 2011;31(4):1489–1499. doi: 10.1523/JNEUROSCI.4134-09.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Astley SJ, Little RE. Maternal marijuana use during lactation and infant development at one year. Neurotoxicol Teratol. 1990;12(2):161–168. doi: 10.1016/0892-0362(90)90129-z. [DOI] [PubMed] [Google Scholar]
- 150.Long JZ, Nomura DK, Vann RE, et al. Dual blockade of FAAH and MAGL identifes behavioral processes regulated by endocannabinoid crosstalk in vivo. Proc Natl Acad Sci USA. 2009;106(48):20270–20275. doi: 10.1073/pnas.0909411106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Straiker A, Mackie K. Depolarization-induced suppression of excitation in murine autaptic hippocampal neurones. J Physiol. 2005;569(Pt 2):501–517. doi: 10.1113/jphysiol.2005.091918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152■■.Wang X, Dow-Edwards D, Anderson V, Minkoff H, Hurd YL. In utero marijuana exposure associated with abnormal amygdala dopamine D2 gene expression in the human fetus. Biol Psychiatry. 2004;56(12):909–915. doi: 10.1016/j.biopsych.2004.10.015. Along with [74], it is one of only two human studies to date that describe changes in neurotransmitter signaling by as early as midgestation in response to PME. [DOI] [PubMed] [Google Scholar]
- 153.Dahl RE, Scher MS, Williamson DE, Robles N, Day N. A longitudinal study of prenatal marijuana use. Effects on sleep and arousal at age 3 years. Arch Pediatr Adolesc Med. 1995;149(2):145–150. doi: 10.1001/archpedi.1995.02170140027004. [DOI] [PubMed] [Google Scholar]
- 154.Chandler LS, Richardson GA, Gallagher JD, Day NL. Prenatal exposure to alcohol and marijuana: effects on motor development of preschool children. Alcohol Clin Exp Res. 1996;20(3):455–461. doi: 10.1111/j.1530-0277.1996.tb01075.x. [DOI] [PubMed] [Google Scholar]
- 155.Fried PA, O'connell CM, Watkinson B. 60- and 72-month follow-up of children prenatally exposed to marijuana, cigarettes, and alcohol: cognitive and language assessment. J Dev Behav Pediatr. 1992;13(6):383–391. [PubMed] [Google Scholar]
- 156.Day NL, Richardson GA, Geva D, Robles N. Alcohol, marijuana, and tobacco: effects of prenatal exposure on offspring growth and morphology at age six. Alcohol Clin Exp Res. 1994;18(4):786–794. doi: 10.1111/j.1530-0277.1994.tb00041.x. [DOI] [PubMed] [Google Scholar]
- 157.Fried PA, James DS, Watkinson B. Growth and pubertal milestones during adolescence in offspring prenatally exposed to cigarettes and marihuana. Neurotoxicol Teratol. 2001;23(5):431–436. doi: 10.1016/s0892-0362(01)00161-1. [DOI] [PubMed] [Google Scholar]
- 158.Willford JA, Chandler LS, Goldschmidt L, Day NL. Effects of prenatal tobacco, alcohol and marijuana exposure on processing speed, visual-motor coordination, and interhemispheric transfer. Neurotoxicol Teratol. 2010;32(6):580–588. 201. doi: 10.1016/j.ntt.2010.06.004. [DOI] [PMC free article] [PubMed]
- 201.Substance Abuse and Mental Health Services Administration. Results from the 2009 National Survey on Drug Use and Health Mental Health Findings (Center for Behavioral Health Statistics and Quality, NSDUH Series H-39, HHS Publication No SMA 10-4609) 2010 www.oas.samhsa.gov/NSDUH/2k9NSDUH/MH/2k9MHResults.htm.
- 202.Substance Abuse and Mental Health Services Administration: Substance Use among Women During Pregnancy and Following Childbirth. The NSDUH Report. 2009 http://oas.samhsa.gov/2k9/135/PregWoSubUse.htm.
- 203.United Nations Office on Drugs and Crime: United Nations Office on Drugs and Crime (UNODC), World Drug Report 2010 (United Nations Publication, Sales No. E.10. XI.13) www.unodc.org/documents/wdr/WDR_2010/World_Drug_Report_2010_lo-res.pdf.