

Abstract
Stem cell function is an exquisitely regulated process. To date, however, the contribution of metabolic cues to stem cell function is poorly understood. Here we identify a novel PML - Peroxisome-proliferator activated receptor delta (PPARδ) - fatty acid oxidation (FAO) pathway for haematopoietic stem cell (HSC) maintenance. We have found that loss of Ppard profoundly affects the maintenance of HSCs. Moreover, treatment with PPARδ agonists improves these HSC functions, whereas, conversely, inhibition of mitochondrial FAO induces loss of the HSC compartment. Importantly, we demonstrate that PML exerts its essential role in HSC maintenance through regulation of PPAR signalling and FAO. Mechanistically, the PML-PPARδ-FAO pathway controls HSC asymmetric division. Depletion of Ppard or Pml, as well as FAO inhibition, results in symmetric commitment of HSC daughter cells while, conversely, PPARδ activation increases asymmetric division. Thus, our findings identify a new metabolic switch for the control of HSC cell fate with important therapeutic implications.
Haematopoietic stem cells (HSCs) are found in a quiescent state in the bone marrow (BM) niche and are the source of all haematological progenitors and differentiated cells throughout organismal lifespan1–6. To define the modes and mechanisms that regulate self-renewal and commitment of stem cells represents one of the central tasks of stem cell biology, since alterations in this equilibrium greatly impact haematopoietic homeostasis and maintenance. It has been suggested that asymmetric division of HSCs ensures that a fraction of the daughter cells retain stem cell features while replenishing the committed compartment7–10. Therefore identifying the factors regulating this process is of great biological and therapeutic relevance.
Peroxisome proliferator-activated receptors (PPARs: PPARα, PPARβ/δ and PPARγ, encoded by PPARA, PPARD and PPARG genes, respectively) are key members of the nuclear receptor super-family of transcription factors, which are in charge of nutrient sensing and the transcriptional regulation of metabolic pathways11, especially fatty acid transport and oxidation (fatty acid oxidation, FAO)12,13. Activators and inhibitors of these pathways have been developed, and PPAR agonists have been tested for the treatment of obesity and metabolic disorders14,15.
The Promyelocytic leukaemia (PML) tumour suppressor gene, originally cloned at the break point of the t(15;17) chromosomal translocation of acute promyelocytic leukaemia (APL), plays a key role in the maintenance of HSCs16. The PML protein is the essential component of the PML nuclear bodies (PML-NBs), sub-nuclear structures that have been implicated in a wide variety of processes including post-translational modifications and transcriptional regulation17. How PML exerts such a critical function in the biology and maintenance of HSCs remains to be established.
Over the past few years, the relationship between HSC function and metabolism has begun to be better understood18–22. Recent studies have focused largely on glycolysis and energy homeostasis, however, while the contribution of lipid catabolism to HSC maintenance has not been defined. In this study, we identify a novel PPAR-FAO pathway that is critical to the maintenance of HSCs and the control of asymmetric cell division. Moreover, we place this metabolic pathway downstream of PML, an essential rheostat for HSC maintenance. Importantly, these observations have straightforward therapeutic implications for both the improvement of bone marrow transplantation (BMT) efficacy and the treatment of haematological malignancies.
Results
Ppard plays a central role in HSC maintenance
The contribution of lipid catabolism to HSC function has not yet been defined. PPARs have been established as critical regulators of the transcriptional program underlying FAO, however. We therefore investigated whether PPAR and FAO could be relevant to HSC biology.
We first analysed the expression levels of the PPAR family in the HSC compartment, and found that Pparδ was the predominant receptor expressed there (Supplementary Figs. S1a, b). We next assessed whether PPARδ function played a role in HSC maintenance. To this end, we conditionally deleted Ppard in KitposScaposLinneg (KSL) cells ex vivo through retroviral Cre transduction and sorting of GFP-positive infected cells, in order to evaluate the characteristics of Ppard-deficient long-term HSCs (CD34negKSL; Fig. 1a). We initially investigated Ppard-deleted HSC function in vivo in the context of bone marrow transplantation (BMT). Upon transplantation, Ppard loss did not affect homing of HSCs (Fig. 1b and Supplementary Fig. S1c), but profoundly impacted long-term repopulating capacity (Fig. 1c, d and Supplementary Fig. S1d). Critically, the defective repopulation capacity of Ppard-deleted HSCs was further evidenced in the second and third rounds of serial BMT (Fig. 1e, f). It is worth noting that no obvious differences in the expression of previously described genes involved in the maintenance of the HSC compartment were observed in Ppard-deleted HSCs (Supplementary Fig. S1e).
Figure 1. PPARδ is essential for HSC maintenance.

(a) Overview of the experimental design for the deletion of Ppard. KSL, c-KitposSca-1posLinneg, MOI, multiplicity of infection, CAFC, cobblestone area forming cells, LTC-IC, long-term culture initiating cells. (b) Homing capacity to the bone marrow (BM) of indicated HSCs. The mean percentages ± S.D. of donor-derived CD34negKSL cells are shown (n = 3). (c,d) Haematopoietic reconstitution capacity of Ppard-deleted KSL cells in BMT. Recipient mice were transplanted with 1.5x103 PpardΔ/Δ KSL cells plus 2.0x105 competitor cells in competition assays. Results represent the mean percentages ± s.d. of donor-derived cells (n = 4) (c) and multi-lineage haematopoietic contribution (myeloid cells (left), B cells (middle) and T cells (right)) 5 months after transplantation (d). (e,f), Limiting-dilution competitive repopulation analysis in second (e) and third (f) BMT with Ppard-ablated HSCs. (g), Cell cycle status of Ppardlox/lox CD150posCD48negKSL cells by Pyronin Y staining 6 weeks after BMT (n = 4). All error bars indicate s.d.
Consistent with these data, deletion of Ppard also led to a dramatic decrease in the function of HSCs in vitro, with reduced cobblestone area-forming cells (CAFC), a surrogate indicator of HSC maintenance (Supplementary Fig. S1f)23 and colony forming ability in long-term culture (defined as long-term culture initiating cell assay, LTC-IC assay; Supplementary Fig. S1g–i), together with a profound reduction in ATP levels (Supplementary Fig. S1j). However, Cre expression itself did not have a significant effect on the proliferation and differentiation of stem/progenitor cells (Supplementary Fig. S1k).
It has been shown that several HSC-regulatory factors modulate the stem cells’ exit from quiescence; therefore, we evaluated the relevance of Ppard for the maintenance of a quiescent state. Interestingly, Ppard-deletion in the HSC resulted in exit from quiescence (Fig. 1g and Supplementary Fig. S1l).
PPARδ activators enhance HSC function
We then tested whether pharmacological PPARδ activation would lead to increased maintenance and function in vivo and in vitro. First, we treated 2-month-old mice with GW501516 (GW) daily for 28 days (Supplementary Fig. S2a), and observed that GW administration did not alter the proliferation, differentiation, or overall number of progenitor cells at the steady state, even as it increased long-term culture initiating cell (LTC-IC) capacity (Supplementary Fig. S2b–d and data not shown).
Next, we employed two different pharmacological activators of PPARδ at low doses in vitro (L165,041; L165 and GW)24–26. As predicted, incubation with PPARδ activators increased the number of cobblestone areas (Supplementary Fig. S2e), in line with the increased expression of PPARδ targets (Supplementary Fig. S2f) and ATP levels (Supplementary Fig. S2g). Critically, we found that culturing HSCs in the presence of PPARδ agonists increased their ability to generate colonies of differentiated haematopoietic cells in LTC-IC assays (Supplementary Fig. S2h, i).
To provide a definitive proof of the potential benefit of PPARδ activators in the HSC compartment, we then tested the effect of GW in BMT. To this end, we transplanted 1.5x103 KSL cells in lethally irradiated mice, and we subjected them to treatment with vehicle or GW daily during the period of HSC homing, lodgement and engraftment to the BM niche27–29 (Fig. 2a). Six weeks after transplantation, when a major donor contribution to haematopoiesis is observed (data not shown), we harvested the BM and analysed the number of remaining HSCs. Treatment with GW significantly increased the number of long-term HSCs in the BM (Fig. 2b and Supplementary Fig. S2j). Notably, similar results were obtained with shorter treatments of 2-weeks with GW (Supplementary Fig. S2k, l). Importantly, GW treatment significantly increased long-term BM and multi-lineage haematopoietic reconstitution capacity after a second BMT of KSL cells or MNCs (Fig. 2c–e).
Figure 2. Pharmacological activation of PPARδ enhances HSC maintenance.
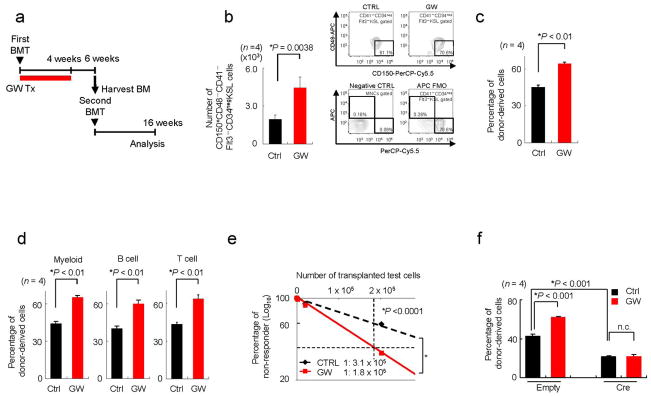
(a) Overview of the experimental design for GW treatment (Tx) in serial BMT. (b) Mean values ± s.d. of donor-derived CD150posCD48negCD41negCD34negFlt3negKSL cells in recipient mice (left panels) and representative flow cytometry data and are shown (right upper panels) (n = 4). Negative Ctrl as well as fluorescence minus one (FMO) for CD48 are also demonstrated as control (right lower panels). (c–e). Donor-derived haematopoietic contribution 16 weeks after second round BMT with donor-derived KSL cells (c, d) or bone marrow mononuclear cells (BMMNCs, e) (as described in (a)) (n = 4). Results represent mean percentages ± s.d. of donor-derived cells in myeloid cells (left), B cells (middle) and T cells (right) in recipient mice 16 weeks after secondary BMT (n = 4) (d). (f) Effect of PPARδ agonist in the repopulation capacity of WT or Ppard-depleted KSL cells in 16 weeks after BMT. All error bars indicate s.d.
In order to demonstrate that PPARδ agonists did exert their beneficial activity through the selective cell autonomous activation of PPARδ in the HSCs, we treated Ppard-Wild type (WT) or Knockout (KO) HSCs with the PPARδ agonist GW. As predicted, Ppard-KO HSCs were refractory to the effect of GW in both CAFC and LTC-IC assays (Supplementary Fig. S3a, b), and the repopulation capacity of KSL cells and MNCs upon BM transplant (Fig. 2f and Supplementary Fig. S3c). Furthermore, in vitro treatment with GW in the absence of stromal cells still increased function of WT stem cells (Supplementary Fig. S4).
Taken together, these data reveal PPARδ as a critical druggable modulator of HSC maintenance and function in a haematopoietic stem cell-autonomous manner.
Inhibition of mitochondrial FAO impairs maintenance of the HSC
PPAR transcription factors are central regulators of nutrient sensing, metabolic reprogramming and differentiation11. In particular, PPARδ, together with PPARα, plays a critical role in the sensing of fatty acids and the activation of the FAO transcriptional program13,30. We therefore evaluated the requirement of active FAO for HSC maintenance.
We first measured FAO in both undifferentiated and differentiated haematopoietic cells. KSL cells exhibited detectable and measurable FAO, which was determined by incubating the cells in the presence or absence of maximal doses of Etomoxir (a pharmacological inhibitor of mitochondrial beta-oxidation of long-chain fatty acids, which does not affect the oxidation of short-chain FA nor peroxisomal FAO, 100μM) in the assay (Fig. 3a)31,32. However, FAO analysis with the same cell number of lineage positive cells did not render a signal that was significantly over the counts obtained upon incubation with Etomoxir (Fig. 3a). Notably, FAO analysis with a greater number of differentiated cells (up to 2.5 fold more Lineage positive cells than KSL cells) yielded similar results (data not shown). This data is consistent with the hypothesis that PPARδ signalling is downregulated during the course of haematopoietic differentiation (Supplementary Fig. S5a).
Figure 3. Pharmacological inhibition of mitochondrial FAO with Etomoxir induces HSC exhaustion.

(a) FAO in undifferentiated (KSL) and differentiated (Lineagepos) cells (n = 3). (b) Effect of etomoxir treatment on transplanted HSC function. After transplantation, recipient mice were treated with vehicle or Etomoxir for 4 weeks and BM was harvested 6 weeks after transplantation. Representative flow cytometry data (right) and mean numbers of donor-derived stem cell compartment in recipient mice (left) 6 weeks after BMT are shown. (c,d) Multi-lineage haematopoietic contribution. Recipient mice were treated with Etomoxir for 2 weeks. Results represent mean percentages of donor-derived cells in mononuclear cells (c) and in the indicated lineages (d) in recipient mice 24 weeks after transplantation. (e,f) Effect of etomoxir on the reconstitutive capacity of HSCs in secondary round BMT, following the experimental design outlined in Fig. 2a. Donor-derived haematopoietic contribution (e) and frequency of HSCs (f, n = 10) were determined 16 weeks after second round BMT. (g,h) Experimental design (g, left) and repopulation capacity of HSCs treated with Etomoxir in second round BMT. Donor-derived haematopoietic contribution (g, right) and functional HSC activity (h, n = 10). All error bars indicate s.d.
To further evaluate whether mitochondrial FAO plays a role in stem cell biology, we first evaluated in vivo the impact of Etomoxir on the ability to repopulate the BM. Notably, we did not observe any toxic effect of Etomoxir in our experimental settings, in line with previous reports33. Four-week treatment with Etomoxir after BMT in lethally irradiated mice led to a significant reduction in long-term HSCs in BM (Fig. 3b). Even a shorter treatment (of 2 weeks) with Etomoxir after BMT resulted in a profound alteration of long-term CD34negFlt3−KSL cells in the BM and donor-derived cells in peripheral blood 24 weeks after transplantation (Fig. 3c,d and Supplementary Fig. S5b). Moreover, we observed reduced long-term BM and haematopoietic reconstitution capacity in secondary BMT with two different protocols of serial BMT (Fig. 3e–h). Importantly, both in vitro and in vivo, the doses of Etomoxir employed in these experiments initially increased the number of total HSCs, but did not affect cell survival, nor did they display toxicity in the HSC compartment (Supplementary Fig. S5c–e).
We also analysed the impact of pharmacological mitochondrial FAO inhibition in vitro. Incubation with Etomoxir led to a reduction in the number of cobblestone areas (Supplementary Fig. 5f). We then tested the impact of Etomoxir treatment on an LTC-IC assay, and found that Etomoxir triggered an opposite response to that observed with PPARδ activators, but similar to that observed in Ppard-deficient cells, i.e., an initial expansion (coherent with increased exit from quiescence) followed by exhaustion of the compartment (Supplementary Fig. S5g–i). In accord with previous data, Etomoxir also led to a reduction in levels of FAO and ATP (Supplementary Fig. S5j, k). Importantly, we confirmed the cell-autonomous effect of Etomoxir on sorted haematopoietic stem cells in vitro (Supplementary Fig. S4).
In order to test whether PPARδ activation-mediated enhancement of HSC function depended on mitochondrial FAO, we treated HSCs with GW, Etomoxir, or both. Interestingly, we observed that inhibition of mitochondrial FAO partially or completely prevented the beneficial effect of GW in vitro and in vivo (Supplementary Fig. S6a–e). We also investigated whether the effect of Etomoxir depended on a functional PPARD. In line with our data, Etomoxir had a reduced effect on HSC maintenance accompanied with ATP reduction in vitro and in vivo in the absence of Ppard (Supplementary Fig. S6f–h).
Taken together, these data uncover a role for FAO downstream of PPARδ in the regulation of HSC maintenance.
PPARδ activation rescues the maintenance defect of Pml-KO HSCs
We have previously shown that deletion of Pml results in loss of HSC maintenance, and consequently exhaustion of the HSC compartment (Supplementary Fig. S7a–d and Ref. 16). Surprisingly, we found that Pml-deficient HSCs exhibit reduced expression of PPARD targets (Fig. 4a; similar results were obtained using 36B4 as a housekeeping endogenous control; data not shown). This observation, together with the notion that PML regulates the activity of several transcriptional factors and cofactors17, led us to hypothesize that regulation of PPAR signaling and FAO by PML in the HSC might be of relevance to its maintenance, and that, in turn, the defect in maintenance observed in Pml-KO HSCs could be rescued by pharmacologically enhancing the activity of PPARδ.
Figure 4. Forced pharmacological activation of PPARδ rescues the maintenance defect of Pml-deficient HSCs.

(a) Relative expression of Cpt1a (left) and Acox1 (right) in Pml−/− CD34negKSL cells compared with Pml+/+ CD34negKSL cells. β-Actin was used as an internal control. (b) FAO in Pml+/+ or Pml−/− in KSL cells. Mean fold change from Pml+/+ KSL cells over Etomoxir values are shown. (c) Overview of the experimental design for GW treatment (Tx) in the serial BMT. (d) Representative flow cytometry (right) and mean numbers of donor-derived CD150posCD48negCD41negCD34negFlt3negKSL cells in recipient mice 6 weeks after BMT. (e) Reconstitutive capacity of Pml−/− HSCs treated with GW in secondary BMT. Donor-derived haematopoietic contribution 16 weeks after second round BMT. (f) CAFC frequencies (in LDA) and LTC-IC ability (relative to vehicle-treated Pml+/+) in Pml+/+ and Pml−/− CD34negKSL cells treated with vehicle (Ctrl), GW or L165 (n = 3–4). All error bars indicate s.d.
We ascertained the contribution of PML to the function of PPAR signaling and FAO in cells from haematopoietic origin. As stated above, Pml-deficient HSCs exhibited reduced expression of PPAR targets (Fig. 4a) and ATP levels (Supplementary Fig. S7e). Mechanistically, we found that PML expression resulted in decreased acetylation of PGC1A, a critical co-factor of PPARs, in line with the notion that PML controls key regulators of the acetylation machinery of PGC1A34 (Supplementary Fig. S7f). Importantly, Pml-deficient HSC exhibited diminished FAO compared to Pml-WT cells (Fig. 4b), whereas no difference was observed in differentiated cells (Supplementary Fig. S7g), in agreement with the thesis that Pml expression is markedly reduced during the course of haematopoietic differentiation.16 Taken together these data suggest that PML regulates PPAR signaling and FAO in the HSC.
In order to evaluate the contribution of PPAR signaling to the defect observed in Pml-deficient cells, and since Pparδ is the main PPAR receptor expressed in this compartment (Fig. 1a and Supplementary Fig. S1a), we tested whether pharmacological activation of Pparδ would rescue the loss of maintenance observed in these cells. We first tested whether PPARδ activators would do so when administered systemically in vivo. As reported, transplantation of Pml-KO HSCs with WT competitor cells in lethally irradiated mice led to the expansion-exhaustion of the Pml-KO compartment (Fig. 4c–e and Supplementary Fig. S8)16,35. Strikingly, treatment of mice with GW for 14 days resulted in the prevention of the expansion phase and the amelioration of exhaustion in the Pml-KO compartment (Supplementary Fig. S8a,b). Consistent with this data, GW led to a significant recovery in the number of Pml-KO HSCs in the BM of recipient mice (Fig. 4c–e and Supplementary Fig. S8c–f). A treatment as short as two weeks after first transplantation in Pml-KO cells significantly increased the number of HSCs in the BM after secondary BMT (Supplementary Fig. S8g–i; see Supplementary Fig. S8a for the experimental design). Importantly, 4-week GW treatment of mice upon transplantation with Pml-KO HSCs (see experimental design in Fig. 4c) rescued the number of resident HSCs in the BM 6 weeks after transplantation, and ameliorated Pml-KO HSC function in secondary transplantation (Fig. 4d,e).
In vitro, PPARδ activators increased cobblestone area formation in Pml-KO HSCs (Fig. 4f and Supplementary Fig. S9a) to a greater extent than in WT HSCs (GW, 1.4 fold in WT versus 3.5 fold in KO and L165, 1.6 fold in WT versus 2.4 fold in KO), in line with the recovery in PPARδ signalling observed in conditions of GW treatment in Pml-KO HSCs (Supplementary Fig. S9b) and a recovery in ATP levels (Supplementary Fig. S9c). Strikingly, PPARδ activators normalized both the expansion and subsequent exhaustion phases observed in Pml-KO HSCs by LTC-IC assay in vitro (Fig. 4g and Supplementary Fig. S7a,b and S9d). These data collectively demonstrate that the defect observed in the Pml-KO HSCs arises, at least in part, from dysfunctional PPARδ signalling and FAO.
Pml-loss results in PPARδ-dependent loss of asymmetric division
We next asked what cellular outcomes might result from the beneficial effects of PML-PPARδ activation and FAO on HSC maintenance. Asymmetric division has recently been suggested as a regulator of cell fate decisions in the mammalian haematopoietic system, playing crucial roles in stem cell renewal7. Therefore we hypothesized that PML regulation of PPARδ signalling and FAO may be essential for asymmetric division.
We studied cell fate decision by utilizing a binary assay which takes advantage of two surface markers: Tie2 and CD482,36. In a purified CD150posCD48negCD41negFlt3negCD34neg KSL population from wild type mice, we observed Tie2 positivity in up to 90% of cells (Fig. 5a). In single cell culture assays, after the first cell division, more than 40% of these cells asymmetrically divided to give rise to two distinct daughter cells (asymmetric division), Tie2posCD48neg (HSC) and Tie2negCD48pos (committed cell), while 20% provided two committed Tie2negCD48pos cells (symmetric commitment; Fig. 5b).
Figure 5. An immunophenotypical assay to characterize the asymmetric division in HSCs.

(a) Tie2 and CD48 expression in 50 randomly selected CD150posCD48negCD41negFlt3negKSL cells (n = 3). (b) Division patterns of Tie2posCD48neg HSCs (100 randomly selected cells, Representative data from n = 3). (c) Functional difference of two daughter cells after an initial cell division of CD150posCD48negCD41negFlt3negCD34negKSL cells was investigated in in vitro paired daughter cell assay by long-term culture initiating cell (LTC-IC) analysis (18 divisions -Total; 36 daughter cells-analysed per experiment, n = 3). (d) In vivo paired daughter cell assay experimental design. Right panel depicts a representative division pattern proportion from 36 divisions. All error bars indicate s.d.
To assess the functional robustness of the assay, we investigated the functional asymmetry of daughter cells after the initial cell division. To this end, we employed a functional analysis of paired daughter cells in vitro by LTC-IC analysis (Fig. 5c). The two daughter cells after an initial cell division of CD150posCD48neg CD41negFlt3negCD34neg KSL cells were separated, and their LTC-IC capacity was analyzed. Consistent with the immunophenotypical Tie2/CD48 characterization, the functional asymmetry, in which one daughter cell holds long-term repopulation capacity but another does not, was observed in more than 40% of divisions (Fig. 5c). Critically, we confirmed the functional asymmetry of daughter cells in vivo in BMT assays, where each daughter cell was transplanted into irradiated mice and its short-and long-term repopulating capacity were evalulated (Fig. 5d and Supplementary Fig. S10a and Supplementary Table 1, 2). Notably, the fact that symmetric divisions towards two long-term (LT)-HSCs are observed in vitro (through immunophenotyping), but not in in vivo functional assay (Supplementary Table 2), suggests that the production of LT-HSCs from a parental cell is a rather infrequent process, although there is potential for its occurrence.
After having validated the assay, we then investigated the effects of PPARδ activation and FAO on the cell fate decision of HSCs. Strikingly, Ppard-deletion resulted in a reduced asymmetric division rate and increased symmetric commitment, while no significant effect of Cre expression was observed in these experiments (Fig. 6a, b). Next, we corroborated the immunophenotypical asymmetric division features with functional analysis, and showed that Ppard-loss resulted in decreased function of paired daughter cells in vitro (Fig. 6c). We then tested whether Etomoxir would similarly affect asymmetric division, and we did indeed observe increased symmetric commitment and decreased asymmetric division in Etomoxir-treated HSCs (Fig. 6d). We validated this in vivo as well as in vitro in paired daughter cell assays (Fig. 6e, Supplementary Fig. S10b, and data not shown). In addition, and in line with our previous results, PPARδ activation with GW increased asymmetric division, an effect that was suppressed by pharmacological inhibition of mitochondrial FAO with Etomoxir (Fig. 6f). A higher dose of Etomoxir (20 μM) had a more profound effect than a lower dose of 10 μM, in line with a further reduction in FAO as well as FAO-derived ATP levels (Supplementary S10c, d), and in turn counteracted more effectively the action of GW. We also investigated the effect of another pharmacological FAO inhibitor, Perhexiline, on the division balance of HSCs. As predicted, Pehexiline treatment increased symmetric commitment, and importantly, counteracted the positive effect of GW treatment on asymmetric division (Supplementary Fig. S10e, f). These data provide a compelling explanation for the loss of maintenance observed in Etomoxir-treated HSCs, placing PPAR signaling and FAO at the regulatory core of asymmetric division and haematopoietic stem cell fate.
Figure 6. Ppard and fatty acid oxidation regulate asymmetric division in the HSC compartment.

(a–b) Expected division patterns (a, left) and division pattern of Ppard+/+ and PpardF/F stem cells infected with empty or Cre vector (n = 3, 50 randomly selected divisions per experiment). Scale bar, 10μm. (c) Functional difference of two daughter cells after an initial cell division of Ppard-ablated cells in in vitro paired daughter cell assay (n = 3, 18 divisions from three mice in each experiment). (d) Division pattern in CD150posCD48neg CD41negFlt3negCD34negKSL cells treated with Etomoxir. Scale bar, 10μm. (e) In vivo paired daughter cell assay upon Etomoxir treatment. Percentage for asymmetric division, in which one daughter cell holds long-term repopulation capacity but another does not, is shown. (f) Single cell deposition of CD150posCD48negCD41negFlt3negCD34negKSL cells from mice treated with GW for one week was conducted followed by in vitro treatment with GW and/or Etomoxir (+; 10 μM, ++; 20 μM). Division pattern in each treatment was investigated (n = 9). (g) Division pattern of WT or Pml−/− HSCs. Scale bar, 10μm. (h) CD48neg cells after first division of Pml−/− HSCs in vivo. Sorted CD150posCD48negCD41neg Flt3negCD34negKSL were stained with CSFE followed by their transplantation. 3 days after BMT, CD48 positivity in cells which undergo one division was determined. (i) Division pattern in Pml−/− HSCs treated with vehicle or GW. (j) A model for regulation of asymmetric division by PML-PPARδ-FAO. All error bars indicate s.d.
On this basis, we then tested whether Pml-loss would similarly result in increased symmetric commitment. As predicted, genetic or pharmacological (upon Arsenic trioxide treatment) depletion of Pml mimicked Ppard-loss and FAO inhibition, resulting in reduced asymmetric division and increased symmetric commitment (Fig. 6g and Supplementary Fig. S10g, h). Critically, the consequences of the aberrant division pattern alteration in HSCs were confirmed in vivo (Fig. 6h and Supplementary Fig. S10i–k). In agreement with our previous observations, the phenotype observed in Pml-KO HSCs was rescued upon treatment with the PPARδ agonist GW (Fig. 6i).
Finally, we evaluated whether the observed increase in symmetric commitment could be responsible for the loss of maintenance observed in vivo in Ppard and Pml-deleted HSCs. To this end, we analysed the fraction of CD48 positive cells within the CD34negKSL fraction (Supplementary Fig. S11). In line with our hypothesis, Ppard and Pml deletion resulted in an initial increase of CD34negKSL cells, with a clear enrichment in the fraction of cells that were positive for CD48. Conversely, GW treatment decreased the fraction of CD48 positive cells within the CD34negKSL compartment of Pml-deficient cells, but had no effect on Ppard-deleted cells. As aforementioned, we have also found that loss of Pml or Ppard results in exit from quiescence without induction of apoptosis (Fig. 1g and Supplementary Fig. S1l, Fig. S5i, Fig. S7a,b, Fig. S10k, Fig. S12, and Ref 16). Therefore functional loss along the PML-PPARδ-FAO axis triggers loss in quiescence combined with excessive commitment, which results in the accumulation of committed HSCs (CD34negCD48posKSL) that fail to maintain haematopoiesis at later stages.
Discussion
Our study uncovers a novel and critical metabolic requirement for HSC maintenance and function. We find that PPARδ signalling and FAO are central players in maintaining this delicate equilibrium. These observations are particularly relevant to stem cell therapy, since both the PPAR pathway and FAO are amenable to pharmacological manipulation. It has been proposed that PPAR activators exert beneficial activities in patients with metabolic disorders37,38, and are exercise mimetics15, which has encouraged the use of this family of compounds as health-state promoting drugs. Moreover, selective PPARδ activators have undergone Phase I and II clinical trials for the treatment of symptoms related to metabolic disorders, such as body fat and dyslipidemia (NCT00388180 and NCT00158899 and Ref. 39), and therefore their safety for human use has been evaluated. The benefit of PPARδ on HSC function increases the interest of PPARδ activators for therapy, especially in conditions where the stem cell function needs to be maximized and the amount of the biological sample is rate-limiting, as is the case with BMT. Notably, although our data highlight a major contribution of FAO downstream PPARδ, we cannot rule out the contribution of additional PPARδ-dependent, FAO-independent pathways in HSC maintenance.
We have previously shown that As2O3, a PML-targeting compound, is highly effective in the induction of cell cycle entry of quiescent LICs.16 In this study, we demonstrate that Pml-deficient HSCs exhibit reduced PPAR signalling and FAO, and that defects in maintenance can be ameliorated by enforcing PPARδ activity. Thus, it is tempting to propose that the use of pharmacological inhibitors of FAO may represent a strategy to target not only the bulk of leukaemic cells40, but also to promote leukaemia-initiating cell (LIC) exhaustion. However, a careful assessment of the possible dosing and regimen schedule is needed before using these compounds in the clinic. Importantly, as described for other LIC-targeting drugs16, a combination of these drugs with compounds targeting the proliferating leukaemic cell pool is likely required to effectively manage the disease.
Pharmacological mTOR inhibition partially rescues the maintenance defect of Pml-deficient HSCs16. Interestingly, inhibition of mTOR by rapamycin has been shown to increase FAO and activate PPAR signalling43 in multiple cell types41,42 and to activate PPAR signalling43, therefore suggesting that PPARδ- and mTOR-dependent pathways downstream of PML might converge in the control of FAO towards HSC maintenance.
It has recently been suggested that haematopoietic cells in mammals can divide asymmetrically as a way to tightly control the decision of self-renewal versus differentiation7,44. Our in vitro and in vivo analyses demonstrate that the fate of HSCs (maintenance vs. exhaustion) is accompanied by profound changes in the rate of asymmetric versus symmetric division. Moreover, we here identify PML-PPARδ-FAO as the first metabolic pathway known to regulate this process, whose impairment results in excessive symmetric commitment (Fig. 6j).
Our findings may also provide a molecular explanation for the haematopoietic alterations observed in telomerase-deficient mice45 and in Lkb1-deleted HSCs20–22, where downregulation in PGC1A expression is observed. On the basis of our data, these effects may in part impinge on components of the PML/PGC1A/PPAR pathway, resulting in impaired FAO and symmetric commitment.
FAO has been shown to be relevant to sustaining ATP levels when breast epithelial cells lose contact with the extracellular matrix46. In an analogous biological process, it is plausible that a functional PML-PPAR-FAO regulatory network ensures the proper execution of an asymmetric HSC division by protecting cells when they lose contact with the bone marrow niche during cell division through the adequate production of ATP. Indeed, recent reports support the idea that a decrease in ATP levels is associated with HSC exhaustion20,21. In line with this hypothesis, we observe that the PML-PPARδ-FAO pathway does sustain ATP levels in the HSC (Supplementary Fig. S1j, Fig. S2g, S5k, Fig. S6h, Fig. S7e, Fig. 9c, Fig. S10d,f). Thus our data lend support to the notion that fatty acid metabolism downstream of PML and PPARD provides the required energy to allow the proper execution of asymmetric division, hence favouring the maintenance and function of the haematopoietic stem cell compartment, although other mechanisms downstream of PPARD are also likely to be relevant to this activity. It must be noted, however, that current technological limitations warrant a direct in vivo imaging of the HSC division process in the years to come.
In summary, this study uncovers a function for PML-PPARδ and FAO in the control of HSC asymmetric division and maintenance, and opens in turn new therapeutic avenues for normal haematopoietic and leukaemic stem cell function.
Online Methods
Mice
Generation of Pml-deficient mice has been described47. C57BL/6 mice (B6-CD45.2) and C57BL/6 mice congenic for the CD45 locus (B6-CD45.1) were purchased from The Jackson Laboratory and crossed with 129Sv mice. F6 B6/Sv129 mice were used as recipients in transplantation assays as described16. GW501516 (5 mg per kg body weight per day; Enzo life sciences) and Etomoxir (25 mg per kg body weight, day ON day OFF; Sigma) were administered by oral gavage and i.p. injections, respectively. The animal protocol was reviewed and approved by the Beth Israel Deaconess Medical Center Institute Animal Care and Use Committee (IACUC). The Ppard floxed allele was generated as described previously48. To delete Ppard in haematopoietic system, MSCV-Puro-IRES-GFP or Puro-IRES-GFP-Cre retroviral vectors were employed in purified KSL cells was utilized49.
Reagents
GW501516 was dissolved for in vivo experiments as previously published33, in Carboxymethylcellulose, and mice where treated with the GW solution or the Carboxymethylcellulose solution alone (vehicle). For in vitro experiments, GW and L165 were dissolved in DMSO, and DMSO was used in vehicle-treated cells. ATP levels in stem cell compartment was analysed by ATP lite luminescence ATP detection kit (Perkin Elmer, Inc., CA) according to manufacturer’s instruction.
Quantitative Real time PCR
Total RNA was extracted from cells using RNeasy kit (Qiagen) including DNAse digestion. cDNA was obtained with First-strand cDNA synthesis kit (USB). Taqman probes were obtained from Applied Biosystems and selected to avoid detection of genomic DNA. Amplifications were run in a 7900 Real-Time PCR System (Applied Biosystems). Each value was adjusted by using 36B4/Rplp0 or β-actin levels as reference.
Western blotting, immunoprecipitation and immunofluorescence
Cells were harvested and protein was extracted as previously described50. Retroviral infection and acetylation experiments were performed as previously described51,52. Briefly, 106 K562 cells were transfected with Nucleofection reagent V (Lonza) using Amaxa nucleoporator, following manufacturers’ instructions (program T-16) and with the DNA ratio indicated above (3.5 μg of total DNA). After 24 hours, cells were lysed in 40 mM Tris (pH 7.6), 150 mM NaCl, 1 mM EDTA, 1 mM MgCl2, 1% Triton X100, 1 mM sodium ortho-vanadate (Na3VO4), 1 mM NaF, 1 mM b-Glycerophosphate, 14 mM Nicotinamide, 400 nM trichostatin and protease inhibitor cocktail (Hoffmann–La Roche), cleared by centrifugation and subjected to immunoprecipitation with Flag-conjugated beads (Sigma) (in the same buffer with 0.1% Triton X100). After 2–3 hours, beads were washed, resuspended in laemmli loading buffer (Boston Bioproducts), boiled and the supernantant was subjected to SDS-PAGE. Proteins were detected using a rabbit polyclonal anti-PML antibody (Bethyl), anti-PGC1A antibody (H-300, Santa Cruz Biotechnology), anti-acetyl lysine (9441, Cell Signalling) and anti-β-actin monoclonal antibody (Sigma).
Fatty acid oxidation
Cells were incubated overnight in culture medium containing 100 μM palmitate (C16:0) and 1 mM carnitine. In the final 2 (MEFs) or 4 (Haematopoietic cells) hours of incubation, cells were pulsed for 4 hours with 1.7 μCi [9,10(n)-3H]palmitic acid (GE Healthcare) in the presence or absence of Etomoxir (100 μM, sigma), and the medium was collected to analyse the released 3H2O, formed during cellular oxidation of [3H]palmitate52,53. Briefly, medium was TCA precipitated, supernatants were neutralized with NaOH and loaded onto ion exchange columns packed with DOWEX 1X2-400 resin (Sigma). The radioactive product was eluted with water and quantitated by liquid scintillation counting. For haematopoietic cells, 70,000 cells (Fig. 3a and Fig. 4b) or 125,000 cells (Supplementary Fig. S7g) were assayed (as counted by sorting; the presence of an equal cell number at the time of the assay was validated by cell counting with haematocytometer for Fig. 4b). Etomoxir, a specific inhibitor of CPT1α, was used to specifically inhibit mitochondrial FAO. Alternatively, to determine FAO in small number of cells, oxygen consumption rate (OCR) immediately following the exposure of HSC to palmitate was used as an indicator of FAO in a XF96 Analyzer (Seahorse Bioscience)54,55, where the appropriateness of the method is presented by the decrease in OCR by the incubation with increasing doses with the CPT1 inhibitor.
Flow cytometry
For flow cytometric analyses, we used monoclonal antibodies specific for the following: CD41 (eBioMWRag30), Flt-3 (Avas12a1), CD34 (RAM34), c-Kit (2B8), Sca-1 (E13-161.7), CD3e (145-2C11), CD4 (L3T4), CD8 (53-6.72), B220 (RA3-6B2), TER-119 (TER-119), Gr-1 (RB6-8C5), CD34 (RAM34), CD11b (M1/70), IgM (II/41), CD19 (eBio1D3) and NK-1.1 (PK136); all were from eBioscience. Anti-CD150 (TC15-12F12.2) and CD48 (HM48-1) antibodies were from BioLegend. We used a mixture of monoclonal antibodies against CD4, CD8, CD3e, B220, TER-119, CD11b, Gr-1, IgM, CD19 and NK-1.1 as a lineage marker (Lineage). We performed Pyronin Y staining as previously described2.
Long-term cultures and colony-forming assays
For long-term cultures, we co-cultured 50 CD34negKSL cells with OP9 cells in Minimum Essential Media, alpha modification (αMEM Sigma) containing 12.5% FCS (JRH Bioscience), 12.5% horse serum (Gibco BRL) and 1 nM dexamethasone. After 2, 4, 8 or 12 weeks of culture, we harvested cells and used them for haematopoietic colony forming assays as described2,16. Frequencies of CAFC and LTC-IC were calculated by limiting-dilution method (maximum likelihood from the proportion of negative wells measured for each input dilution of cells tested)47. In each experiment, 36 (CAFC) and 12 (LTC-IC) replicates of each dilution were performed, and a split-plot ANOVA analysis is utilized to investigate the difference between two conditions. For treatment experiments, 0.1 μM GW501516 (Enzo life sciences), 0.5 μM L165,041 (Sigma), 10 or 20 μM Etomoxir (Sigma), or 2 μM Perhexiline (Sigma) was added to cultures.
Competitive reconstitution assay
We sorted 1.5 x 103 KSL cells from wild-type or Pml−/− mice (CD45.2). We transplanted cells into lethally irradiated CD45.1 congenic mice in competition with 2 x 105 bone marrow mononuclear cells from CD45.1 mice. We monitored reconstitution of donor (CD45.2) cells by staining blood cells with antibodies against CD45.2 and CD45.1. For treatment experiments, recipient mice were administered with GW501516 by oral gavage (everyday) and Etomoxir i.p. (every other day). For serial transplantation, donor-derived bone marrow mononuclear cells or KSL cells were collected from recipient mice 6, 12 or 24 weeks after BMT (first BMT) and transplanted into recipient mice with fresh competitor cells (second BMT). Calculation of competitive reconstitution unit (CRU) was conducted using L-Calc software (StemCell Technologies.
Asymmetric division and Paired daughter cell assays
Single cell deposition of CD150posCD48negCD41negFlt3negCD34negKSL cells from 8-week old mice (total 160–320 cells/experiment) were directly performed onto retronectin-coated slides and cells were culture in StemSPAN (StemCell Technologies) supplemented with 50 ng/ml stem cell factor (SCF) and 50 ng/ml thrombopoietin (TPO). Cells were either maintained untreated or treated with vehicle, 0.1 μM GW501516 (Enzo life sciences) and/or 10 μM Etomoxir (Sigma) or 0.15 μM As2O3 (Sigma). Close to 50% of the cells (49.7±5.7 in 100 randomly selected cells) undergo initial cell division at day 3. Cells were stained with anti-mouse Tie2 antibody, anti-mouse CD48 antibody and DAPI at day 3. Student’s t test (as indicated in the figure) was utilized to determine statistical significance, and statistical significance in division pattern as well as the disturbance of division pattern were also confirmed by log-linear model and Chi-square test (data not shown). For paired daughter cell assay, the corresponding daughter cells derived from a single cell were separated followed by LTC-IC assay or BMT assay as previously described56. Single daughter cell is transplanted into lethally irradiated recipient mice in a competitive assay. Long-term repopulation and short-term repopulation are achieved when more than 1% of haematological contribution in peripheral blood of recipient mice is observed, 16 weeks and 4 weeks after BMT respectively57.
Homing analysis
Lethally irradiated Ly45.1 mice were transplanted with 2,000 CD34negKSL cells from PpardF/F mice (Ly45.2) infected with Empty or Cre vector and 2x105 competitor BMMNCs. Cells from the BM of recipient mice were analysed 24 hours post-transplantation to identify their derivation.
Statistical Analysis
Student’s t test was utilized to determine statistical significance unless otherwise specified. P values lower than 0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
We thank all members of the Pandolfi laboratory for comments and discussion. We are especially thankful to Kentaro Hosokawa for technical advice on asymmetric division, to Meritxell Alberich-Jorda and Ana M. Zubiaga for technical help and critical discussions, and Pere Puigserver and Marcia C. Haigis for technical support on metabolic analysis. K.I. was supported by a K99 NIH grant. The work of A.C. was supported by EMBO, the Ramón y Cajal award (Spanish Ministry of Education), ISCIII (PI10/01484), the Marie Curie Reintegration grant (277043) and the Basque Government (PI2012-03). This work was supported by NIH grants to P.P.P.
Footnotes
Author Contributions
K.I., A.C. and P.P.P. conceived and designed the experiments. K.I., A.C. and D.W. performed the experiments. F.A. and T.S. contributed to the setup of the asymmetric division experiments. U.A. performed the statistical analysis. Z.T.S. helped with the approach of ATP measurement. R.M.E. and C.H.L. provided Ppard conditional KO mice. Data was analysed by K.I., A.C. and P.P.P. The paper was written by K.I., A.C. and P.P.P.
Competing interest statement
The authors declare no competing financial interests.
References
- 1.Ergen AV, Goodell MA. Mechanisms of hematopoietic stem cell aging. Exp Gerontol. 2010;45:286–290. doi: 10.1016/j.exger.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arai F, et al. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell. 2004;118:149–161. doi: 10.1016/j.cell.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 3.Fuchs E, Tumbar T, Guasch G. Socializing with the neighbors: stem cells and their niche. Cell. 2004;116:769–778. doi: 10.1016/s0092-8674(04)00255-7. [DOI] [PubMed] [Google Scholar]
- 4.Lemischka IR, Moore KA. Stem cells: interactive niches. Nature. 2003;425:778–779. doi: 10.1038/425778a. [DOI] [PubMed] [Google Scholar]
- 5.Morrison SJ, Spradling AC. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell. 2008;132:598–611. doi: 10.1016/j.cell.2008.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilson A, Trumpp A. Bone-marrow haematopoietic-stem-cell niches. Nat Rev Immunol. 2006;6:93–106. doi: 10.1038/nri1779. [DOI] [PubMed] [Google Scholar]
- 7.Morrison SJ, Kimble J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature. 2006;441:1068–1074. doi: 10.1038/nature04956. [DOI] [PubMed] [Google Scholar]
- 8.Lansdorp PM. Self-renewal of stem cells. Biol Blood Marrow Transplant. 1997;3:171–178. [PubMed] [Google Scholar]
- 9.Metcalf D. Cell-cell signalling in the regulation of blood cell formation and function. Immunol Cell Biol. 1998;76:441–447. doi: 10.1046/j.1440-1711.1998.00761.x. [DOI] [PubMed] [Google Scholar]
- 10.Ogawa M. Stochastic model revisited. Int J Hematol. 1999;69:2–5. [PubMed] [Google Scholar]
- 11.Michalik L, et al. International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol Rev. 2006;58:726–741. doi: 10.1124/pr.58.4.5. [DOI] [PubMed] [Google Scholar]
- 12.Braissant O, Foufelle F, Scotto C, Dauca M, Wahli W. Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology. 1996;137:354–366. doi: 10.1210/endo.137.1.8536636. [DOI] [PubMed] [Google Scholar]
- 13.Takahashi S, Tanaka T, Sakai J. New therapeutic target for metabolic syndrome: PPARdelta. Endocr J. 2007;54:347–357. doi: 10.1507/endocrj.kr-99. [DOI] [PubMed] [Google Scholar]
- 14.Clapham JC, Arch JR. Thermogenic and metabolic antiobesity drugs: rationale and opportunities. Diabetes, obesity & metabolism. 2007;9:259–275. doi: 10.1111/j.1463-1326.2006.00608.x. [DOI] [PubMed] [Google Scholar]
- 15.Narkar VA, et al. AMPK and PPARdelta agonists are exercise mimetics. Cell. 2008;134:405–415. doi: 10.1016/j.cell.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ito K, et al. PML targeting eradicates quiescent leukaemia-initiating cells. Nature. 2008;453:1072–1078. doi: 10.1038/nature07016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bernardi R, Pandolfi PP. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol. 2007;8:1006–1016. doi: 10.1038/nrm2277. [DOI] [PubMed] [Google Scholar]
- 18.Simsek T, et al. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell. 2010;7:380–390. doi: 10.1016/j.stem.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takubo K, et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell. 2010;7:391–402. doi: 10.1016/j.stem.2010.06.020. [DOI] [PubMed] [Google Scholar]
- 20.Gan B, et al. Lkb1 regulates quiescence and metabolic homeostasis of haematopoietic stem cells. Nature. 2011;468:701–704. doi: 10.1038/nature09595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gurumurthy S, et al. The Lkb1 metabolic sensor maintains haematopoietic stem cell survival. Nature. 2011;468:659–663. doi: 10.1038/nature09572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakada D, Saunders TL, Morrison SJ. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature. 2011;468:653–658. doi: 10.1038/nature09571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Breems DA, Blokland EA, Neben S, Ploemacher RE. Frequency analysis of human primitive haematopoietic stem cell subsets using a cobblestone area forming cell assay. Leukemia. 1994;8:1095–1104. [PubMed] [Google Scholar]
- 24.Peters JM, Gonzalez FJ. Sorting out the functional role(s) of peroxisome proliferator-activated receptor-beta/delta (PPARbeta/delta) in cell proliferation and cancer. Biochimica et biophysica acta. 2009;1796:230–241. doi: 10.1016/j.bbcan.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tanaka T, et al. Activation of peroxisome proliferator-activated receptor delta induces fatty acid beta-oxidation in skeletal muscle and attenuates metabolic syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:15924–15929. doi: 10.1073/pnas.0306981100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wurch T, Junquero D, Delhon A, Pauwels J. Pharmacological analysis of wild-type alpha, gamma and delta subtypes of the human peroxisome proliferator-activated receptor. Naunyn Schmiedebergs Arch Pharmacol. 2002;365:133–140. doi: 10.1007/s00210-001-0504-z. [DOI] [PubMed] [Google Scholar]
- 27.Adams GB, et al. Stem cell engraftment at the endosteal niche is specified by the calcium-sensing receptor. Nature. 2006;439:599–603. doi: 10.1038/nature04247. [DOI] [PubMed] [Google Scholar]
- 28.Lapidot T, Dar A, Kollet O. How do stem cells find their way home? Blood. 2005;106:1901–1910. doi: 10.1182/blood-2005-04-1417. [DOI] [PubMed] [Google Scholar]
- 29.Wolf NS. Dissecting the hematopoietic microenvironment. I. Stem cell lodgment and commitment, and the proliferation and differentiation of erythropoietic descendants in the S1-S1d mouse. Cell Tissue Kinet. 1974;7:89–98. doi: 10.1111/j.1365-2184.1974.tb00402.x. [DOI] [PubMed] [Google Scholar]
- 30.Wang YX. PPARs: diverse regulators in energy metabolism and metabolic diseases. Cell research. 2010;20:124–137. doi: 10.1038/cr.2010.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tutwiler GF, Brentzel HJ, Kiorpes TC. Inhibition of mitochondrial carnitine palmitoyl transferase A in vivo with methyl 2-tetradecylglycidate (methyl palmoxirate) and its relationship to ketonemia and glycemia. Proc Soc Exp Biol Med. 1985;178:288–296. doi: 10.3181/00379727-178-42012. [DOI] [PubMed] [Google Scholar]
- 32.Wolf HP. Possible new therapeutic approach in diabetes mellitus by inhibition of carnitine palmitoyltransferase 1 (CPT1) Horm Metab Res Suppl. 1992;26:62–67. [PubMed] [Google Scholar]
- 33.Samudio I, et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest. 2010;120:142–156. doi: 10.1172/JCI38942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kitamura YI, et al. FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metab. 2005;2:153–163. doi: 10.1016/j.cmet.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 35.Carlesso N, Aster JC, Sklar J, Scadden DT. Notch1-induced delay of human hematopoietic progenitor cell differentiation is associated with altered cell cycle kinetics. Blood. 1999;93:838–848. [PubMed] [Google Scholar]
- 36.Kiel MJ, Yilmaz OH, Iwashita T, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 37.Berger JP, Akiyama TE, Meinke PT. PPARs: therapeutic targets for metabolic disease. Trends Pharmacol Sci. 2005;26:244–251. doi: 10.1016/j.tips.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 38.Wagner KD, Wagner N. Peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) acts as regulator of metabolism linked to multiple cellular functions. Pharmacol Ther. 2010;125:423–435. doi: 10.1016/j.pharmthera.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 39.Riserus U, et al. Activation of peroxisome proliferator-activated receptor (PPAR)delta promotes reversal of multiple metabolic abnormalities, reduces oxidative stress, and increases fatty acid oxidation in moderately obese men. Diabetes. 2008;57:332–339. doi: 10.2337/db07-1318. [DOI] [PubMed] [Google Scholar]
- 40.Samudio I, Fiegl M, Andreeff M. Mitochondrial uncoupling and the Warburg effect: molecular basis for the reprogramming of cancer cell metabolism. Cancer Res. 2009;69:2163–2166. doi: 10.1158/0008-5472.CAN-08-3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brown NF, Stefanovic-Racic M, Sipula IJ, Perdomo G. The mammalian target of rapamycin regulates lipid metabolism in primary cultures of rat hepatocytes. Metabolism. 2007;56:1500–1507. doi: 10.1016/j.metabol.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 42.Sipula IJ, Brown NF, Perdomo G. Rapamycin-mediated inhibition of mammalian target of rapamycin in skeletal muscle cells reduces glucose utilization and increases fatty acid oxidation. Metabolism. 2006;55:1637–1644. doi: 10.1016/j.metabol.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 43.Sengupta S, Peterson TR, Laplante M, Oh S, Sabatini DM. mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature. 2011;468:1100–1104. doi: 10.1038/nature09584. [DOI] [PubMed] [Google Scholar]
- 44.Wu M, et al. Imaging hematopoietic precursor division in real time. Cell Stem Cell. 2007;1:541–554. doi: 10.1016/j.stem.2007.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sahin E, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470:359–365. doi: 10.1038/nature09787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schafer ZT, et al. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature. 2009;461:109–113. doi: 10.1038/nature08268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang ZG, et al. Role of PML in cell growth and the retinoic acid pathway. Science. 1998;279:1547–1551. doi: 10.1126/science.279.5356.1547. [DOI] [PubMed] [Google Scholar]
- 48.Barak Y, et al. Effects of peroxisome proliferator-activated receptor delta on placentation, adiposity, and colorectal cancer. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:303–308. doi: 10.1073/pnas.012610299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chambers SM, et al. Hematopoietic fingerprints: an expression database of stem cells and their progeny. Cell Stem Cell. 2007;1:578–591. doi: 10.1016/j.stem.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alimonti A, et al. Subtle variations in Pten dose determine cancer susceptibility. Nat Genet. 2010;42:454–458. doi: 10.1038/ng.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen Z, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gerhart-Hines Z, et al. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. 2007;26:1913–1923. doi: 10.1038/sj.emboj.7601633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Deberardinis RJ, Lum JJ, Thompson CB. Phosphatidylinositol 3-kinase-dependent modulation of carnitine palmitoyltransferase 1A expression regulates lipid metabolism during hematopoietic cell growth. J Biol Chem. 2006;281:37372–37380. doi: 10.1074/jbc.M608372200. [DOI] [PubMed] [Google Scholar]
- 54.Pike LS, Smift AL, Croteau NJ, Ferrick DA, Wu M. Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochimica et biophysica acta. 2010 doi: 10.1016/j.bbabio.2010.10.022. [DOI] [PubMed] [Google Scholar]
- 55.Ferrick DA, Neilson A, Beeson C. Advances in measuring cellular bioenergetics using extracellular flux. Drug Discov Today. 2008;13:268–274. doi: 10.1016/j.drudis.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 56.Iwama A, et al. Enhanced self-renewal of hematopoietic stem cells mediated by the polycomb gene product Bmi-1. Immunity. 2004;21:843–851. doi: 10.1016/j.immuni.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 57.Matsuzaki Y, Kinjo K, Mulligan RC, Okano H. Unexpectedly efficient homing capacity of purified murine hematopoietic stem cells. Immunity. 2004;20:87–93. doi: 10.1016/s1074-7613(03)00354-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.