

Abstract
MED12 is a member of the large Mediator complex, which has a critical and central role in RNA polymerase II transcription. As a multiprotien complex, Mediator regulates signals involved in cell growth, development and differentiation, and it is involved in a protein network required for extraneuronal gene silencing and also functions as a direct suppressor of Gli3-dependent Sonic hedgehog signaling. This may may explain its role in several different X-linked intellectual disability syndromes that share some overlapping clinical features. This review will compare and contrast four different clinical conditions that have been associated with different mutations in MED12, which is located at Xq13. To date, these conditions include Opitz–Kaveggia (FG) syndrome, Lujan syndrome, Ohdo syndrome (Maat-Kievit-Brunner type, or OSMKB), and one large family with profound X-linked intellectual disability due to a novel c.5898insC frameshift mutation that unlike the other 3 syndromes, resulted in affected female carriers and truncation of the MED12 protein. It is likely that more MED12 mutations will be detected in sporadic patients and X-linked families with intellectual disability and dysmorphic features as exome sequencing becomes more commonly utilized, and this overview of MED12-related disorders may help to correlate MED12 genotypes with clinical findings.
Keywords: FG syndrome, Opitz-Kaveggia syndrome, Lujan-Fryns syndrome, MED12, X-linked intellectual disability, Ohdo syndrome (Maat-Kievit-Brunner type)
INTRODUCTION
MED12 encodes a subunit of the macromolecular complex known as Mediator, which is required for thyroid hormone–dependent activation and repression of transcription by RNA polymerase II. The MED12 protein is not part of the Mediator core complex, but is a member of a Mediator module that may act as an adaptor for specific transcription factors. There are more than 25 subunits in the Mediator complex and in 2007, Risheg et al., reported on the first intellectual disability syndrome (FG syndrome, also called Opitz-Kaveggia syndrome) to be linked to a recurrent missense mutation (c.2881C>T, p. R961W) in MED12 at Xq13. This syndrome was initially described by Opitz and Kaveggia [1974] in a family of five affected males with intellectual disability, macrocephaly, imperforate anus and hypotonia. Later in 2007, Schwartz et al., reported a different novel missense mutation (c.3020A>G, p.N1007S) in the MED12 gene in the original family with Lujan syndrome and in a second family that was initially considered to have Opitz–Kaveggia (FG) syndrome. Although Lujan syndrome had not been previously considered to be part of the differential diagnosis of FG syndrome, there were some overlapping clinical manifestations, such as dysgenesis of the corpus callosum, macrocephaly, a tall forehead, hypotonia, intellectual disability, and behavioral disturbances. Thus, these two X-linked intellectual disability syndromesare allelic, with mutations in the MED12 gene.
More recently, novel missense mutations in MED12 have been linked to Ohdo syndrome (Maat-Kievit-Brunner type, OSMKB), which differs from other blepharophimosis-intellectual disability syndromes by X-linked inheritance and facial coarsening at an older age [Vulto-van Silfhout et al., 2013]. Finally, a large family with profound X-linked intellectual disability was recently found to carry a novel c.5898insC frameshift mutation in MED12 through parallel sequencing of all X-chromosome exons [Lesca et al., 2013]. Dysmorphic features in affected males included narrow face, high forehead, flat malar area, high nasal bridge and short philtrum. Language was absent or very limited, but most affected individuals had a friendly personality with occasional aggressive outbursts. Unlike the other conditions with missense mutations in MED12, variable cognitive impairment was noted in seven heterozygous females, with no correlation between cognitive function and X-chromosome inactivation profiles in blood cells. With the increasing use of exome sequencing, it is likely that previously undiagnosed patients with intellectual disability will be found to have mutations in MED12, and the clinical features of such patients can be compared with the recognized phenotypes that are currently associated with mutations in MED12 as described below..
OPITZ-KAVEGGIA (FG) SYNDROME
FG syndrome (Opitz-Kaveggia syndrome, OMIM 305450) was delineated by Opitz and Kaveggia in 1974 based on the clinical findings in three brothers and two of their male first cousins (Table I). In this family, FG syndrome was defined as a multiple congenital anomaly syndrome characterized by relative macrocephaly, broad and flat thumbs, imperforate anus, hypotonia and moderately severe intellectual disability [Opitz and Kaveggia 1974]. Facial characteristics included a prominent forehead, upswept frontal hairline, downslanting palpebral fissures, ocular hypertelorism, and small prominent ears with a simplified helical pattern. The corpus callosum was deficient or absent altogether, with occasional EEG abnormalities. Associated defects included anal stenosis or other malformations of the intestinal tract and heart, hernias, and craniosynostosis, and this combination of defects was sometimes lethal during early childhood. Skeletal manifestations included stature in the lower range of normal, broad and flat thumbs and halluces, partial syndactyly, pectus excavatum, joint contractures, and spinal curvature. Surviving males had congenital hypotonia with constipation, and during early childhood they were friendly, inquisitive, and hyperactive with a very short attention span, while older males were noted to have temper tantrums with attacks of screaming and aggressive or self-abusive behaviors requiring medication with tranquilizers [Opitz and Kaveggia, 1974]. Female carriers were unaffected. A subsequent family with FG syndrome was reported by Keller et al. [1976]. They also noted the distinctive affable, outgoing personality in surviving males. Later reports by McCardle and Wilson [1993]; and Graham et al. [1998] (Family 1 and Family 3) confirmed the distinctive features of FG syndrome associated with the recurrent missense mutation (c.2881C>T, p.R961W) in MED12.
Table 1.
Comparison of Clinical Findings in MED12-Related Disorders
| Major distinguishing findings | Lujan syndrome | FG syndrome | p.S1967Qfsx84a | OSMKBb |
|---|---|---|---|---|
| Growth | ||||
| Tall stature | + | − | − | − |
| Macrocephaly | + | + | − | − |
| Facies | ||||
| Long narrow face | + | − | + | − |
| Triangular face | − | − | − | + |
| Tall prominent forehead | + | + | + | + |
| Facial coarsening | − | − | − | + |
| Hypertelorism/telecanthus | − | + | − | − |
| Strabismus | + | + | − | + |
| Blepharophimosis | − | − | − | + |
| Downslanting palpebrae | + | + | − | + |
| Thick alae nasi | − | − | − | + |
| High nasal root | + | − | + | − |
| High narrow palate | + | + | − | + |
| Dental crowding | + | + | − | − |
| Maxillary hypoplasia | + | + | + | − |
| Micrognathia/retrognathia | + | + | − | + |
| Open mouth | + | + | + | + |
| Frontal hair upsweep | − | + | − | − |
| Small ears | − | + | − | + |
| Hand | ||||
| Long hyperextensible digits | + | − | − | + |
| Broad thumbs | + | + | − | − |
| Persistent fetal finger pads | − | + | − | − |
| Horizontal palmar creases | − | + | − | − |
| Syndactyly | − | + | − | − |
| Neuromuscular | ||||
| Agenesis of corpus callosum | + | + | − | − |
| Congenital hypotonia | + | + | − | + |
| GI | ||||
| Anal anomalies | − | + | − | − |
| Genitourinary problems | − | + | − | − |
| Chronic constipation | − | + | − | + |
| Speech | ||||
| Little or no language | − | − | + | + |
| Hypernasal voice | + | − | − | − |
| Other | ||||
| Intellectual disability | + | + | + | + |
| Behavioral disturbances | + | + | + | + |
| Affected females | − | − | + | − |
From Lesca et al. [2013 From Lesca et al. [in press]
Prior to the discovery of the genetic basis for FG syndrome in 2007 [Risheg et al., 2007], there had been a wide expansion of the range of clinical manifestations associated with FG syndrome, which included the core manifestations of hypotonia, constipation, and short stature with relative macrocephaly, which are relatively common manifestations among persons with cognitive disability. Genetic heterogeneity was suggested by the identification of five candidate loci for FG syndrome, based on the expanded phenotype, which had been mapped to the X chromosome. The existence of genetic heterogeneity was further supported by the identification of mutations in three X-linked genes, FLNA, BRWD3 and UPF3B [Field et al., 2007; Tarpey et al., 2007; Unger et al., 2007] in families with features that appeared to overlap this expanded view of FG syndrome. Accordingly, efforts were made to explore the genetic basis for 30 patients with a previous clinical diagnosis of FG syndrome [Lyons et al., 2009]. One 7-year-old male with mild intellectual disability, hypotonia, and constipation was the only individual recognized as having a phenotype consistent with the original FGS family and other similar families with the MED12 p.R961W mutation. His characteristic facial features included small and simple ears, tall and prominent forehead, frontal hair upsweep, down slanting palpebral fissures, broad thumbs and halluces, fetal fingertips pads, and characteristic friendly behavior, and the recurrent R961W mutation in the MED12 gene was identified.
Three patients were thought to fall within the broader FG syndrome phenotypic spectrum, while the remaining 26 patients presented with a range of clinical features commonly seen in individuals with intellectual disability of unclear etiology [Lyons et al., 2009]. MED12 testing and testing of BRWD3 and UPF3Bwere negative in these 26patients and the three patients thought to fall within the broader FGS phenotypic spectrum. ATRX syndrome was confirmed by identification of an ATRX gene mutation (p.N1860S) in an 11-year-old male with intellectual disability, hypotonia, dysmorphic facial features, and constipation. Multiplex ligation dependent probe amplification (MLPA) analysis of the MECP2 gene confirmed a large heterozygous deletion consistent with Rett syndrome in a 4-year-old female with hypotonia, developmental regression, broad thumbs and halluces, and normal MECP2 mutation analysis. An unbalanced chromosome translocation was suspected in two brothers with disparate clinical phenotypes (deletion of 9q34 and duplication of 22q13 in one brother, and deletion of 22q13 and duplication of 9q34 in the other brother). Chromosomal microarray analysis identified a microdeletion or microduplication in four other patients (2q37.3 deletion, 17p11.2 duplication, 18q22 deletion and 10qter duplication). The significance of the 10q terminal duplication was uncertain, since the same duplication was identified in the patient’s unaffected mother. DHPLC screening of all exons and intron–exon boundaries of the FLNA gene revealed different missense variants in three patients, which had not been previously reported, so their significance was uncertain. These findings emphasize the importance of chromosomal microarrays in the initial evaluation of patients with intellectual disability, combined with genotyping based on careful phenotyping
There is a consistent behavior phenotype in males with FG syndrome and this syndrome should be considered in males who present with developmental delay and a behavioral phenotype of hyperactivity, affability, and excessive talkativeness along with the distinctive clinical features of FG syndrome [Graham et al., 1999; Risheg et al., 2007; Clark et al., 2009; Graham et al., 2010]. Males with the recurrent R961W mutation inMED12have strengths in socialization and daily living skills, despite their communicative deficits [Graham et al., 2008], and their strengths in socialization skills may mask their communicative deficits. Although parents may report that boys with FG syndrome are social and talk excessively, they may have problems in articulation, pragmatic language use, syntax, and intonation, which may be further complicated by their low facial muscle tone and oral–motor discoordination. These language deficits may lead to secondary behavioral problems and difficulties in performing routine daily living skills, such as toilet training, feeding, or dressing. Males with FG syndrome commonly have increased frustration and attention problems, which may also be related to their poor expressive language skills. Thus, early and ongoing language therapy is essential in all males with FG syndrome. Males with FG syndrome can have significant externalizing behaviors and are at increased risk for aggressive behavior because of their deficits in communication skills [Graham et al., 2008].
Since most males with FG syndrome have poor GI motility and constipation throughout their lifetimes, gastrointestinal sources should be sought for unexplained pain including gastroesophageal reflux, constipation and hemorrhoids. Hypotonia and joint laxity, which are common in FG syndrome, can also cause joint misalignment and pain. Headaches, insomnia, and sleep apnea are also common and may exacerbate maladaptive behaviors. Anxiety in males with the FG syndrome appears to be heightened by changes in routines and transitions. Thus, it is important to provide a highly structured environment for males with FG syndrome, since they prefer routines. Providing frequent explicit warnings of transitions and changes in their routines will lessen anxiety, improve coping, and reduce maladaptive behaviors. Hyperactivity may subside during adulthood, when withdrawal and anxiety may become more prominent. They may benefit from mood stabilizers, and their transition to a community living situation can be challenging without careful planning and timely behavioral intervention.
Currently, 10 families with FG syndrome and the recurrent p.R961W mutation in MED12 have been reported (including a total of 23 affected males), and diagnostic criteria have been proposed [Clark et al., 2009]. A family history of X-linked mental retardation, deceased male infants and/or multiple fetal losses was documented in all families. FG syndrome affects the corpus callosum more often than any other organ. Therefore, brain imaging is helpful in evaluating patients suspected to have FG syndrome. Facial features are distinctive, especially the narrow, long face, tall forehead, full upper eyelids and open mouth [Figure 1]. Small, simple prominent ears are a separate criterion because this is almost universal in patients with the recurrentMED12 mutation, and uncommon in mutation-negative patients. The affable personality is also very common in patients with the recurrentMED12 mutation, and uncommon in mutation-negative patients, whereas the presence of anxiety is less discriminatory. Exclusion criteria include female sex, lack of intellectual disability and autosomal patterns of inheritance. One family who met these criteria was found to carry a novel p.G958E mutation in MED12, with mild manifestations in carrier females [Rump et al., 2011].
Figure 1.
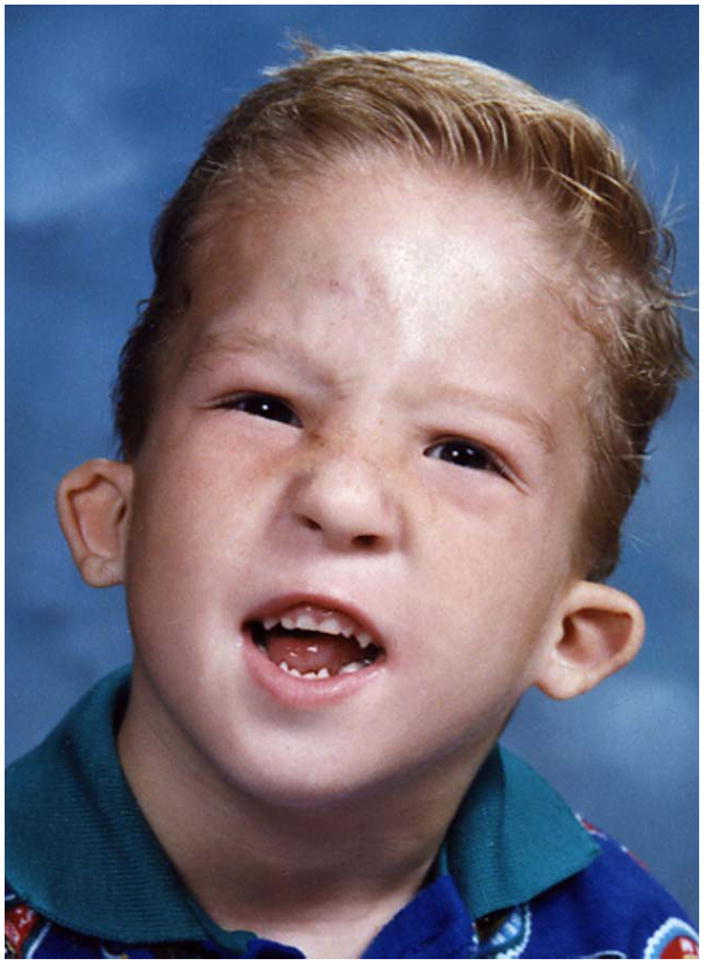
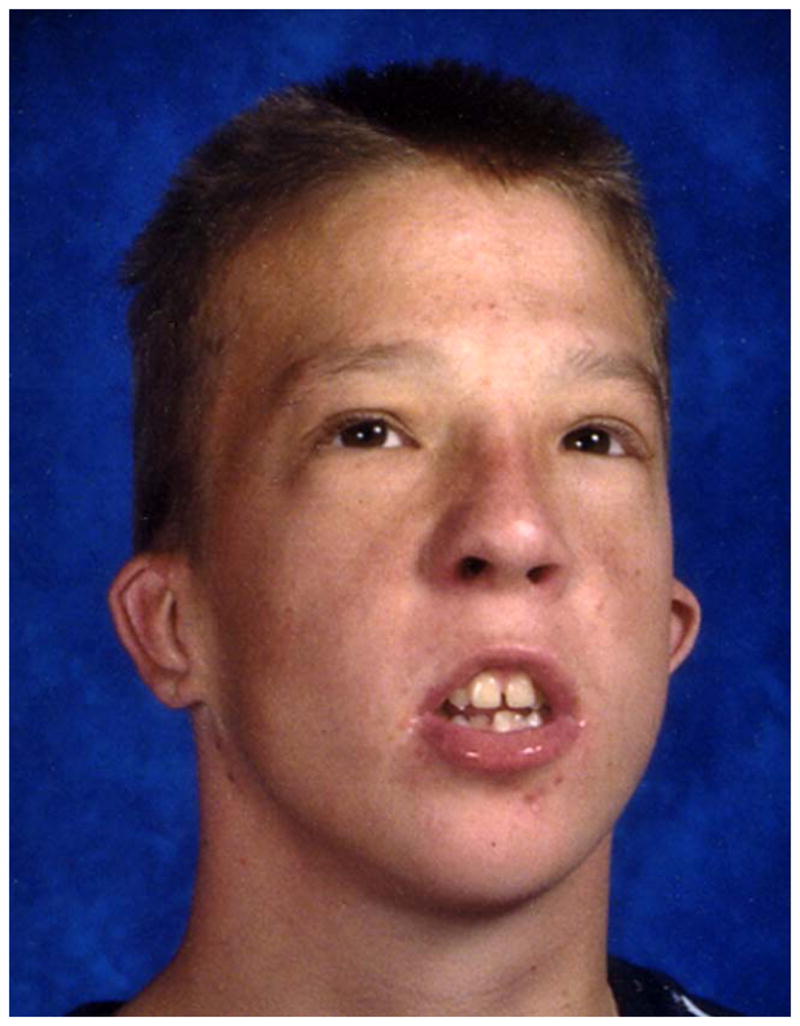
FG syndrome facial features from Case 1 from Graham et al. [1998] shown as a child (A) and in late adolescence (B).
LUJAN SYNDROME
The original family with Lujan syndrome (also known as Lujan–Fryns syndrome and XLID with Marfanoid features) as well as a second unrelated family were found to have a different sequence alteration (c.3020A>G, p.N1007S) in exon 22 of the MED12 gene [Schwartz et al., 2007]. This observation means FG syndrome and Lujan syndrome are allelic, with different mutations in the MED12 gene. Individuals with Lujan syndrome have tall stature with asthenic build, macrocephaly, a tall narrow face, maxillary hypoplasia, a high narrow palate with dental crowding, a small or receding chin [Fig 2], long hands with hyper extensible digits, hypernasal speech, hypotonia, mild-to-moderate intellectual disability, behavioral abnormalities (hyperactivity, emotional lability, shyness, aggressiveness, autistic mannerisms and/or psychoses) and dysgenesis of the corpus callosum [Table I]. The spectrum of behavioral disturbances, ranges from shyness to frank psychosis, while some individuals, seem to be friendly, jovial and outgoing, without overt signs of behavioral disturbances. For those individuals demonstrating overlapping behavioral features with FG syndrome, see the previous section for suggestions on how to manage such behavioral features.
Figure 2.
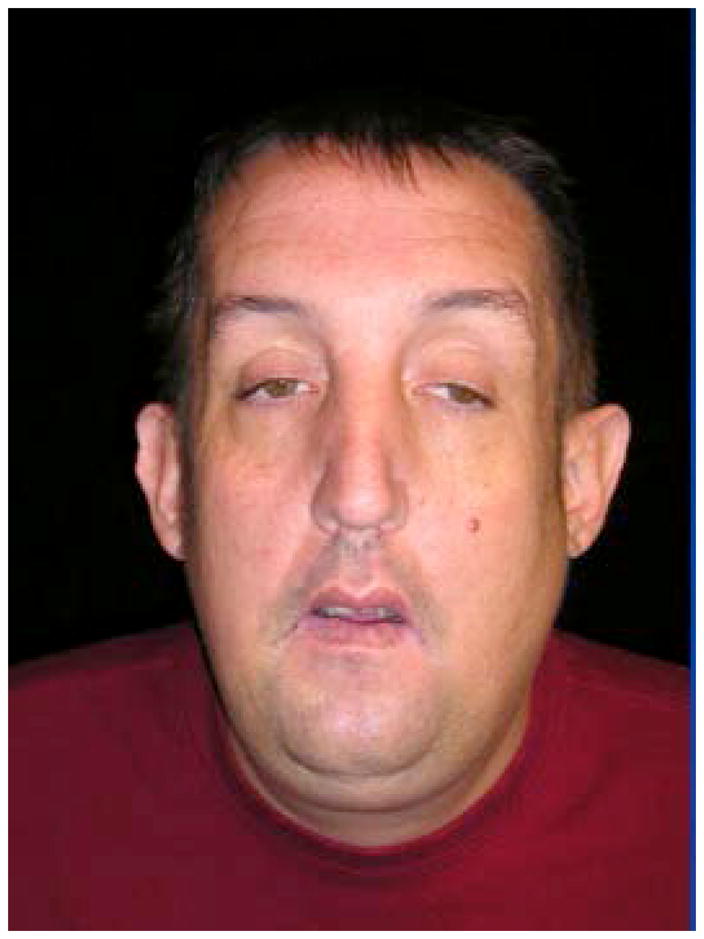
Lujan syndrome facial features in adult from original family in Schwartz et al. [2007].
X-LINKED OHDO SYNDROME (MAAT-KIEVIT-BRUNNER TYPE)
X-linked Ohdo syndrome (Maat-Kievit-Brunner type, or OSMKB) syndrome was initially distinguished from other types of blepharophimosis-intellectual disability syndromes by occurrence in a maternal uncle-nephew pair that seemed compatible with X-linked inheritance [Verloes et al., 2006]. Ohdo syndrome is actually a heterogeneous group of disorders characterized by intellectual disability and distinctive facial features, which include blepharophimosis, ptosis, a round face with a characteristic nose, and a small mouth (Table I, Fig 3.). The blepharophimosis-ID syndromes have been classified into five distinct subgroups by Verloes et al. [2006]. The first group can be distinguished from the others because it is caused by chromosome 3pter deletions. The second group is designated as Ohdo type, and it is characterized prognathism, short philtrum, and proteinuria, with normal growth, muscle tone and limbs. The Verloes type manifests severe microcephaly, epilepsy, brain malformations, adducted thumbs, and genital abnormalities. The Say/Barber/Biesecker/Young/Simpson (SBBYS) type is characterized by a bulbous nasal tip, small abnormal helices, full cheeks, retrognathia, hypotonia, hyper extensible joints, cryptor chidism, and this syndrome is caused by mutations in lysine acetyltransferase 6B (KAT6B). Young males with X-linked OSMKB type resemble Ohdo blepharophimosis syndrome, while older males have coarse facial features, thick alaenasi, and a triangular face [Verloes et al., 2006]. Exome sequencing was performed in two families with OSMKB type. In these two families, MED12 missense mutations (c.3443G>A, p.R1148H or c.3493T>C p.S1165P) segregated with the phenotype. Subsequent analysis of a cohort of nine sporadic males with Ohdo syndrome, revealed one additional patient with a de novo missense change (c.5185C>A p.H1729N) in MED12 [Vulto-van Silfhouet et al., 2013]. The occurrence of three different hemizygous missense mutations in three unrelated families affected by OSMKB type confirmed that mutations in MED12are the cause of this condition.
Figure 3.
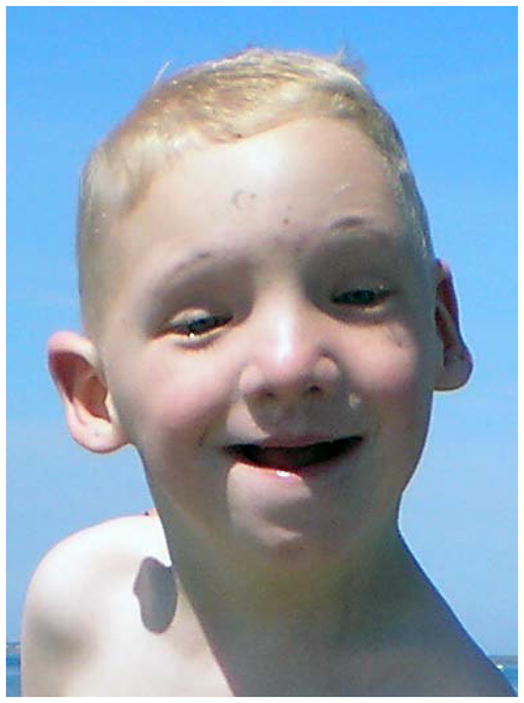
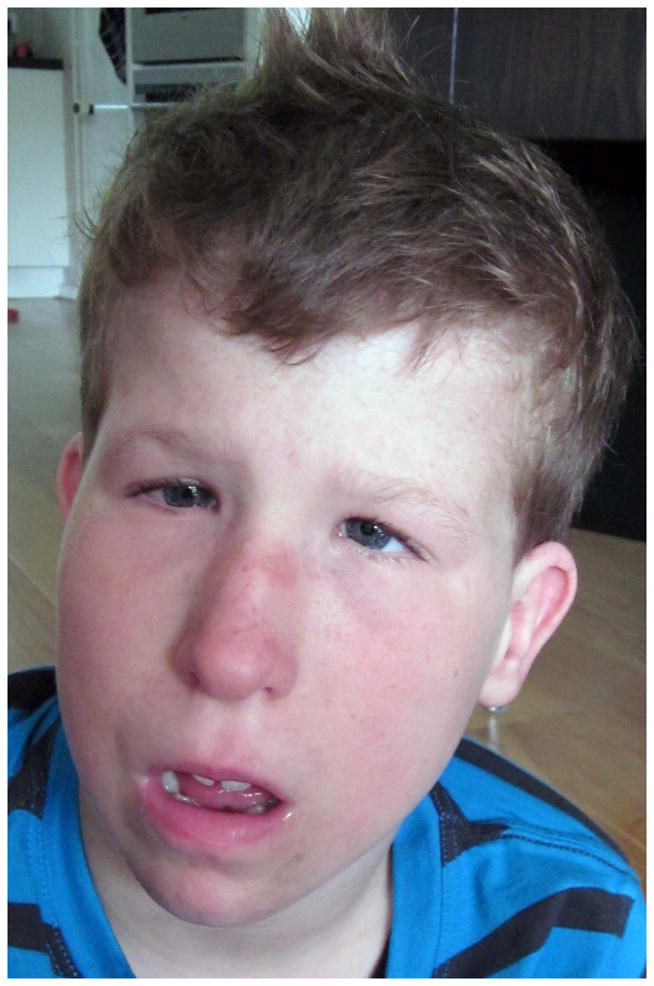
Facial features in OSMKB syndrome in child (A) from Family 1 and adolescent (B) from Family 2 reported by Vulto-van Silfhout [2013] courtesy Anneka T. Vulto-van Silfhout and Han Brunner.
OTHER MED12 PHENOTYPES
Recently, parallel sequencing of all X-chromosome exons identified a novel c.5898insC frameshift mutation (p.S1967Qfsx84) of MED12 in a five-generation family with non-syndromic profound X-linked intellectual disability in 10 males and one female [Lesca et al., 2013]. Clinical features common to most affected males were long narrow face, high forehead, flat malar area, high nasal bridge, short philtrum, and absent or severely-limited language (Table I). Most patients had a friendly personality but some had aggressive outbursts. Cognitive impairment varied from borderline to profound ID in seven heterozygous females, with no correlation between cognitive function and X-chromosome inactivation profiles in lymphocytes. The severe degree of cognitive dysfunction in male patients, with associated cognitive impairment in heterozygous females, suggested the truncating mutations in this family had a more severe effect on MED12 function than the missense mutations, which have previously been reported in other MED12-related syndromes.
DISCUSSION
It is likely that more MED12 mutations will be detected through exome sequencing or whole genome sequencing in sporadic patients or X-linked families with intellectual disability and dysmorphic features as these techniques become more commonly utilized. This overview of MED12-related disorders may help to correlate clinical findings in such patients with sequence variants found in MED12. MED12 is a member of the large Mediator complex, which has a critical and central role in RNA polymerase II transcription. As a multiprotien complex, it regulates signals involved in cell growth, development and differentiation [Conaway et al., 2005]. Recently, Mediator was shown to be involved in a protein network required for extraneuronal gene silencing. The network included G9a histone methyltransferase and REST (RE1 silencing transcription factor) [Ding et al., 2008]. Additionally, the mutations in MED12, which have been associated with both the FG and Lujan syndromes, disrupted MED12’s REST co-repressor function. This finding linked MED12 to neuronal gene expression, and these mutations to the etiology of the XLID syndromes.
TheseMED12 mutations explain more than the cognitive dysfunction in these XLID syndromes. MED12 functions as a direct suppressor of Gli3-dependent Sonic hedgehog signaling [Zhou et al., 2006]. The FG and Lujan MED12 mutations disrupt this suppression, thereby leading to enhanced Sonic hedgehog pathway activation [Zhou et al., 2012]. This dysregulation of Gli3-dependent Sonic hedgehog signaling may contribute to some of the phenotypic features of males with FG and Lujan syndromes listed in Table I. Other MED12 mutations may eventually be shown to have effects on REST function and Gli-3 - dependent Sonic hedgehog signaling.
MED12 appears to play a role in the development of several cancers [Barbieri et al., 2012; Chiang and Oliva, 2013; Matsubara et al., 2013; Rieker et al., 2013]. It should not be surprising that somatic mutations in MED12 might lead to cancer since it is a hub gene interacting with various signaling pathways [Lehner et al., 2006], and it promotes epigenetic silencing of selected genes [Ding et al. 2008]. This is also consistent with the observation that the protein at the germ line level does not tolerate mutations, based on a lack of SNPs in dbSNP and the 1000 Genomes database.
Kaya et al. [2012] reported a novel Xq12q13.3 duplication in an extended family with three affected individuals with microcephaly, mild ventriculomegaly, seizures and autism with intellectual disability. Clinically normal mothers were completely skewed in favor of the normal X chromosome. This Xq12q13.3 duplication was suggested to have its effect by increased dosage of the genes contained in this duplication, which included NLGN3, OPHN1, AR, EFNB1, TAF1, GJB1, and MED12, In response to this report, Prontara et al. [2012] indicated they had seen 2 brothers with a similar phenotype and genomic duplication. Because so many of the genes in this genomic duplication are involved in autism. it is difficult to highlight MED12 as being any more significant that the other genes, but these reports may suggest dysregulation of MED12 and other genes in the same region may have significant effects on neurodevelopment.
Acknowledgments
Supported, in part, by a grant from NIHCD (HD026202-15) and a grant from the South Carolina Department of Disabilities and Special Needs (SCDDSN). Dedicated to the memory of Ethan Francis Schwartz, 1996–1998.
References
- Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van Allen E, Stransky N, Nickerson E, Chae SS, Boysen G, Auclair D, Onofrio RC, Park K, Kitabayashi N, MacDonald TY, Sheikh K, Vuong T, Guiducci C, Cibulskis K, Sivachenko A, Carter SL, Saksena G, Voet D, Hussain WM, Ramos AH, Winckler W, Redman MC, Ardlie K, Tewari AK, Mosquera JM, Rupp N, Wild PJ, Moch H, Morrissey C, Nelson PS, Kantoff PW, Gabriel SB, Golub TR, Meyerson M, Lander ES, Getz G, Rubin MA, Garraway LA. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44(6):685–9. doi: 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang S, Oliva E. Recent developments in uterine mesenchymal neoplasms. Histopathology. 2013;62(1):124–137. doi: 10.1111/his.12048. [DOI] [PubMed] [Google Scholar]
- Clark RD, Graham JM, Jr, Friez MJ, Hoo JJ, Jones KL, McKeown C, Moeschler JB, Raymond FL, Rogers RC, Schwartz CE, Battaglia A, Lyons MJ, Stevenson RE. FG syndrome, an X-linked multiple congenital anomaly syndrome: the clinical phenotype and an algorithm for diagnostic testing. Genet Med. 2009;11:769–75. doi: 10.1097/GIM.0b013e3181bd3d90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conaway RC, Sato S, Tomomori-Sato C, Yao T, Conaway JW. The mammalian Mediator complex and its role in transcriptional regulation. Trends in Biochemical Sciences. 2005;30:250–255. doi: 10.1016/j.tibs.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Ding N, Zhou H, Esteve PO, Chin HG, Kim S, Xu X, Joseph SM, Friez MJ, Schwartz CE, Pradhan S, Boyer TG. Mediator links epigenetic silencing of neuronal gene expression with X-linked mental retardation. Mol Cell. 2008;31:347–359. doi: 10.1016/j.molcel.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field M, Tarpey PS, Smith R, Edkins S, O’Meara S, Stevens C, Tofts C, Teague J, Butler A, Dicks E, Barthorpe S, Buck G, Cole J, Gray K, Halliday K, Hills K, Jenkinson A, Jones D, Menzies A, Mironenko T, Perry J, Raine K, Richardson D, Shepherd R, Small A, Varian J, West S, Widaa S, Mallya U, Wooster R, Moon J, Luo Y, Hughes H, Shaw M, Friend KL, Corbett M, Turner G, Partington M, Mulley J, Bobrow M, Schwartz C, Stevenson R, Gecz J, Stratton MR, Futreal PA, Raymond F. Mutations in the BRWD3 gene cause X-linked mental retardation associated with macrocephaly. Am J Hum Genet. 2007;81:367–374. doi: 10.1086/520677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham JM, Jr, Clark RD, Moeschler JB, Rogers RC. Behavioral features in young adults with FG syndrome (Opitz-Kaveggia syndrome) American Journal of Medical Genetics. 2010;154C:477–485. doi: 10.1002/ajmg.c.30284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham JM, Jr, Tackels D, Dibbern K, Superneau D, Rogers C, Corning K, Schwartz CE. FG syndrome: report of three new families with linkage to Xq12-q22.1. Am J Med Genet. 1998;80:145–156. [PubMed] [Google Scholar]
- Graham JM, Jr, Visootsak J, Dykens E, Huddleston L, Clark RD, Jones KL, Moeschler JB, Opitz JM, Morford J, Simensen R, Rogers RC, Schwartz CE, Friez MJ, Stevenson RE. Behavioral features in 10 patients with FG syndrome (Opitz-Kaveggia syndrome) and the p.R961W mutation in the MED12 gene. American Journal of Medical Genetics. 2008;146A:3011–3017. doi: 10.1002/ajmg.a.32553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaya N, Colak D, Albakheet A, Al-Owain M, Abu-Dheim N, Al-Younes B, Al-Zahrani J, Mukaddes NM, Dervent A, Al-Dosari N, Al-Odaib A, Kayaalp IV, Al-Sayed M, Al-Hassnan Z, Nester MJ, Al-Dosari M, Al-Dhalaan H, Chedrawi A, Gunoz H, Karakas B, Sakati N, Alkuraya FS, Gascon GG, Ozand PT. A novel X-linked disorder with developmental delay and autistic features. Ann Neurol. 2012;71(4):498–508. doi: 10.1002/ana.22673. [DOI] [PubMed] [Google Scholar]
- Keller MA, Jones KL, Nyhan WL, Franke U, Dixson B. A new syndrome of mental deficiency with craniofacial, limb, and anal abnormalities. J Pediatr. 1976;88:589–591. doi: 10.1016/s0022-3476(76)80012-1. [DOI] [PubMed] [Google Scholar]
- Lehner B, Crombie C, Tischler J, Fortunato A, Fraser AG. Systematic mapping of genetic interactions in Caenorhabditis elegans identifies common modifiers of diverse signaling pathways. Nat Genet. 2006;38(8):896–903. doi: 10.1038/ng1844. [DOI] [PubMed] [Google Scholar]
- Lesca G, Moizard M-P, Gerald Bussy G, Boggio D, Hu H, Haas S, Ropers H-H, Kalscheuer VM, Des Portes V, Labalme A, Sanlaville D, Edery P, Raynaud M, Lespinasse J. Clinical and neurocognitive characterization of a family with a novel MED12 gene frameshift mutation. Am J Med Genet. 2013 doi: 10.1002/ajmg.a.36162. in press. [DOI] [PubMed] [Google Scholar]
- Lujan JE, Carlin ME, Lubs HA. A form of X-linked mental retardation with marfanoid habitus. Am J Med Genet. 1984;17:311–322. doi: 10.1002/ajmg.1320170124. [DOI] [PubMed] [Google Scholar]
- Lyons MJ, Graham JM, Jr, Neri G, Hunter AG, Clark RD, Rogers RC, Moscarda M, Boccuto L, Simensen R, Dodd J, Robertson S, DuPont BR, Friez MJ, Schwartz CE, Stevenson RE. Clinical experience in the evaluation of 30 patients with a prior diagnosis of FG syndrome. J Med Genet. 2009;46:9–13. doi: 10.1136/jmg.2008.060509. [DOI] [PubMed] [Google Scholar]
- Matsubara A, Sekine S, Yoshida M, Yoshida A, Taniguchi H, Kushima R, Tsuda H, Kanai Y. Prevalence of MED12 mutations in uterine and extrauterine smooth muscle tumours. Histopathology. 2013;62(4):657–661. doi: 10.1111/his.12039. [DOI] [PubMed] [Google Scholar]
- McCardle P, Wilson B. Language and development in FG syndrome with callosal agenesis. J Commun Disord. 1993;26:83–100. doi: 10.1016/0021-9924(93)90002-r. [DOI] [PubMed] [Google Scholar]
- Opitz JM, Kaveggia EG. The FG syndrome: an X-linked recessive syndrome of multiple congenital anomalies and mental retardation. Z Kinderheilk. 1974;117:1–18. doi: 10.1007/BF00439020. [DOI] [PubMed] [Google Scholar]
- Prontera P, Ottaviani V, Isidori I, Stangoni G, Donti E. Xq12-q13.3 duplication: evidence of a recurrent syndrome. Ann Neurol. 2012;72(5):821–822. doi: 10.1002/ana.23754. author reply 822–823. [DOI] [PubMed] [Google Scholar]
- Rieker RJ, Agaimy A, Moskalev EA, Hebele S, Hein A, Mehlhorn G, Beckmann MW, Hartmann A, Haller F. Mutation status of the mediator complex subunit 12 (MED12) in uterine leiomyomas and concurrent/metachronous multifocal peritoneal smooth muscle nodules (leiomyomatosis peritonealis disseminata) Pathology. 2013;45(4):388–392. doi: 10.1097/PAT.0b013e328360bf97. [DOI] [PubMed] [Google Scholar]
- Risheg H, Graham JM, Jr, Clark RD, Rogers RC, Opitz JM, Moeschler JB, Peiffer AP, May M, Joseph SM, Jones JR, Stevenson RE, Schwartz CE, Friez MJ. A recurrent mutation in MED12 leading to R961W causes Opitz-Kaveggia syndrome. Nature Genetics. 2007;39:451–453. doi: 10.1038/ng1992. [DOI] [PubMed] [Google Scholar]
- Rump P, Niessen RC, Verbruggen KT, Brouwer OF, de Raad M, Hordijk R. Anovelmutation in MED12 causes FG syndrome (Opitz-Kaveggia syndrome) Clin Genet. 2011;79:183–188. doi: 10.1111/j.1399-0004.2010.01449.x. [DOI] [PubMed] [Google Scholar]
- Schwartz CE, Tarpey PS, Lubs HA, Verloes A, May MM, Risheg H, Friez MJ, Futreal PA, Edkins S, Teague J, Briault S, Skinner C, Bauer-Carlin A, Simensen RJ, Joseph SM, Jones JR, Gecz J, Stratton MR, Raymond FL, Stevenson RE. The original Lujansyndrome family has a novel missense mutation (p.N1007S) in the MED12 gene. J Med Genet. 2007;44:472–477. doi: 10.1136/jmg.2006.048637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarpey PS, Raymond FL, Nguyen LS, Rodriguez J, Hackett A, Vandeleur L, Smith R, Shoubridge C, Edkins S, Stevens C, O’Meara S, Tofts C, Barthorpe S, Buck G, Cole J, Halliday K, Hills K, Jones D, Mironenko T, Perry J, Varian J, West S, Widaa S, Teague J, Dicks E, Butler A, Menzies A, Richardson D, Jenkinson A, Shepherd R, Raine K, Moon J, Luo Y, Parnau J, Bhat SS, Gardner A, Corbett M, Brooks D, Thomas P, Parkinson-Lawrence E, Porteous ME, Warner JP, Sanderson T, Pearson P, Simensen RJ, Skinner C, Hoganson G, Superneau D, Wooster R, Bobrow M, Turner G, Stevenson RE, Schwartz CE, Futreal PA, Srivastava AK, Stratton MR, Gécz J. Mutations in UPF3B, a member of the nonsense-mediated mRNA decay complex, cause syndromic and nonsyndromic mental retardation. Nat Genet. 2007;39:1127–1133. doi: 10.1038/ng2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger S, Mainberger A, Spitz C, Bähr A, Zeschnigk C, Zabel B, Superti-Furga A, Morris-Rosendahl DJ. Filamin A mutation is one cause of FG syndrome. Am J Med Genet A. 2007;143A:1876–1879. doi: 10.1002/ajmg.a.31751. [DOI] [PubMed] [Google Scholar]
- Verloes A, Bremond-Gignac D, Isidor B, David A, Baumann C, Leroy MA, Stevens R, Gillerot Y, Héron D, Héron B, Benzacken B, Lacombe D, Brunner H, Bitoun P. Blepharophimosis-mental retardation (BMR) syndromes: A proposed clinical classification of the so-called Ohdo syndrome, and delineation of two new BMR syndromes, one X-linked and one autosomal recessive. Am J Med Genet A. 2006;140:1285–1296. doi: 10.1002/ajmg.a.31270. [DOI] [PubMed] [Google Scholar]
- Vulto-van Silfhout AT, de Vries BB, van Bon BW, Hoischen A, Ruiterkamp-Versteeg M, Gilissen C, Gao F, van Zwam M, Harteveld CL, van Essen AJ, Hamel BC, Kleefstra T, Willemsen MA, Yntema HG, van Bokhoven H, Brunner HG, Boyer TG, de Brouwer AP. Mutations in MED12cause X-Linked Ohdo Syndrome. Am J Hum Genet. 2013;92:401–406. doi: 10.1016/j.ajhg.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Kim S, Ishii S, Boyer TG. Mediator modulates Gli3-dependent Sonic hedgehog signaling. Mol Cell Biol. 2006;26:8667–8682. doi: 10.1128/MCB.00443-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Spaeth JM, Kim NH, Xu X, Friez MJ, Schwartz CE, Boyer TG. MED12 mutations link intellectual disability syndromes with dysregulated GLI3-dependent Sonic Hedgehog signaling. Proc Natl Acad Sci U S A. 2012;109:19763–19768. doi: 10.1073/pnas.1121120109. [DOI] [PMC free article] [PubMed] [Google Scholar]