
Endothelium-Derived Relaxing Factors and Endothelial Function: A Systematic Review
 , , and
, , and
Abstract
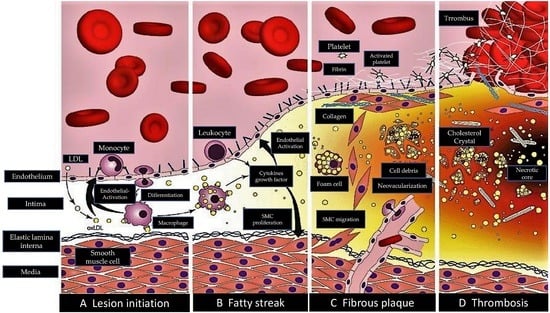
:
1. Introduction
2. Methods
2.1. Search Strategy
2.2. Study Selection and Data Extraction
2.3. Endpoints and Effect Summary
3. Results
3.1. Reactive Oxygen Species

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| First Author/Year Ref | Type of Study | Cohort | Aims | Findings |
|---|---|---|---|---|
| Wang et al. 2017 JAHA [22] | Rat Model | SHR WKY Control | TRPC3 induced ROS production induced. | Improved TRPC3 activity at the cytoplasmic and mitochondrial levels. Increased redox signaling and calcium dysregulation. |
| Montezano et al. 2016 JAHA [23] | Mice model | SM22+, Nox5+, Nox5+/SM22+ † WT | Nox5 in pro-contractile signaling and vascular function. | Nox5 lead to join calcium and reactive oxygen species to the pro-contractile molecular apparatus in vascular smooth muscle cells. |
| Ikumi et al. 2020 J Cardiovasc Pharmacol [24] | Mice model | eNOS-KO nNOS/eNOS-double-KO WT control | EDH in coronary microcirculation and cardiac diastolic function. | Substantial effect of EDH/H2O2 in maintaining coronary microcirculation and cardiac diastolic function through oxidative PKGIα activation. |
| Denhartig et al. 2017 Arterioscler Thromb Vasc Biol [25] | Mice model | C57BL/6 NOX4 deletion C57BL/6 control | NOX4-derived ROS in the development of obesity and insulin resistance. | Substantial role of NOX4-derived ROS in the onset of insulin resistance and adipose tissue inflammation. |
| Sano et al. 2017 Nutr Metab Cardiovasc Dis [26] | Mice model | C57BL/6 HFD C57BL/6 HFD plus EC | In vivo effects of EC on adipose tissue inflammation and obesity. | Noticable beneficial effects of EC for the prevention of adipose tissue inflammation and insulin resistance by marked suppression of CCL19. |
| Li et al. 2022 [27] PLoS One | Rat Model | AT1aR−/− gene KO WT control | The role of Ang II in diet-induced obesity. | AT1aR deficiency alleviated adipocyte hypertrophy in high-fat diet rats by promoting adipose lipolysis probably via cAMP/PKA pathway. |
| La Favor et al. 2016 Arterioscler Thromb Vasc Biol [28] | Human model | Men 15 Women 27 BMI 21.6 ± 0.6 30.1 ± 0.4 36.6 ± 0.7 ℷ | Determination the impact of in vivo ROS on microvascular endothelial function in obese human individuals. | Substantial increase activity of NADPH oxidase. Excessive ROS production in skeletal muscle of obese individuals. Association between excessive NADPH oxidase-derived ROS to microvascular endothelial dysfunction in obesity. |
| Masi et al. 2018 Arterioscler Thromb Vasc Biol [29] | Human model | 36 obeses 31 controls | Impact of arginase as a determinant of endothelial dysfunction in small arteries in obese individuals. | Arginase lead to microvascular endothelial dysfunction in obesity. Decreased effect of arginase in aged obese for higher levels of vascular oxidative stress. Accelerated microvascular remodeling in obese individuals. |
| Godo et al. 2016 Arterioscler Thromb Vasc Biol [30] | Mice model | Cav-1-KO eNOS-Tg WT | Study the probable relevance of the physiological balance between NO and EDH in cardiovascular homeostasis. | Genotypes showed altered cardiovascular phenotypes, including cardiac hypertrophy in Cav-1-KO mice and hypotension in eNOS-Tg mice. Evidence suggested that excessive endothelium-derived NO with reduced EDH impairs cardiovascular homeostasis in mice in vivo. |
| Gray et al. 2016 Arterioscler Thromb Vasc Biol [31] | Human model Mouse model | HAECs_ diabetic HAECs_ non diabetic | Investigate the role of type 4 isoform (NOX4) in human and mouse atherosclerosis. | Both in humans and in mouse, the H2O2 -forming NOX4, in contrast with the superoxide-forming NOX1, can act as a negative modulator of inflammation and remodeling and convey atheroprotection. |
| Otsuka et al. 2013 Ann Cardiothorac Surg [32] | Human model | Comparative study IMA vs. SVG | IMA reveals fewer fenestrations, lower intercellular junction permeability. Higher antithrombotic production of heparin sulfate and tissue plasminogen activator, and NO. | IMA is the first line of defense for the treatment of coronary artery disease. |
| Freed et al. 2014 Circ Res [33] | Human model | Human healthy adipose arterioles pretreated with ceramide | To evaluate the induction of ceramide to favor a switch from nitric oxide to H2O2. | Ceramide has an crucial role in the transition from nitric oxide to mitochondrial-derived H2O2. |
| Durand et at 2016 Arterioscler Thromb Vasc Biol [34] | Human model | Patients with CAD | To evaluate vascular actions of angiotensin 1-7 (ANG 1-7) in human atrial and adipose arterioles. | ANG 1-7 treatment is sufficient to restore the NO component of FMD in arterioles from patients with CAD. |
| Kirsch et al. 2016 Arterioscler Thromb Vasc Biol [35] | Mouse model | Mouse Cremaster vs. Txnrd2-deficient mice | To examine dysregulated redox homeostasis and inadequate nitric oxide signaling. | Txnrd2 plays a crucial important role in balancing mitochondrial ROS production in the endothelium. |
| Erb et al. 2016 Arterioscler Thromb Vasc Biol [36] | Human model | Patients (n = 51) undergoing CABG with IMA | To evaluate both polymorphisms of nitric eNOS gene in the promoter (T-786C) and exon 7 (G894T). | Observed eNOS polymorphisms in larges conduits. Deteriorated endothelium-dependent vasodilatory capacity in patients with CAD. |
3.2. Shear Stress
| First Author/Year Ref | Type of Study | Cohort | Aims | Findings |
|---|---|---|---|---|
| Shindo et al. 2016 Arterioscler Thromb Vasc Biol [51] | Mouse model | Mouse Lipus Vs Mouse non Lipus | LIPUS implicated in amelioration of LV remodeling after IMA. Elucidate the underlying molecular mechanisms involved in the beneficial effects of LIPUS. | LIPUS therapy ameliorates post-myocardial infarction LV remodeling in mice in vivo. Increased vascular endothelial growth factor signaling |
| Hatanaka et al. 2016 Am J Physiol Cell Physiol [52] | Human model | HUVECs Vs HUVECs Knockdown of caveolin-1 or β1-integrin | Effects of SW irradiation on intracellular signaling pathways in vitro to induce myocardial angiogenesis | Activation of caveolin-1 and β1-integrin, and subsequent phosphorylation of Erk and Akt play crucial roles in the SW-induced angiogenesis. |
| Friederich-Persson et al. 2017 Arterioscler Thromb Vasc Biol [53] | Rat model | Wild-type Nox4−/− | Regulatory role and vasoprotective effects of BAT | BAT, via Nox4-derived hydrogen peroxide, induces cyclic GMP-dependent protein kinase G type-1α activation, resulting in reduced vascular contractility |
| Burgoyne et al. 2007 Science [54] | Mice model | SM22+, Nox5+, Nox5+/SM22+ † WT | Whether concentration of oxydants in cells can regulate biochemical signaling mechanisms | Oxydants lead to cGMP-independent vasorelaxation in the cardiovascular system. H2O2 can operate as an endothelium-derived hyperpolarizing factor |
| Prysyazhna et al. 2012 Nat Med. 2012 [55] | Mice model | WT KI | Importance of PKGI-α oxidation in the EDHF mechanism and blood pressure control in vivo | C42S ‘redox-dead’ version of PKGI-α blocked the vasodilatory action of H2O2 on resistance vessels resulting in hypertension in vivo. |
| Noblet et al. 2015 Arterioscler Thromb Vasc Biol [56] | Suine model | Ossabaw swine obese vs. lean | Effects of lean and obese coronary PVAT on coronary vasodilation | Lean and obese coronary PVAT attenuates vasodilation via inhibitory effects on vascular smooth muscle K (+) channels. Calpastatin initiate or lead to progression of smooth muscle dysfunction in obesity. |
| Dou et al. 2017 Arterioscler Thromb Vasc [57] | Human model | AT-RAA (N = 74) AT-Mediastianal (n = 74) | AT-expressed ADAM17 activation in development of coronary microvascular dysfunction in obesity. | Aging and obesity decrease caveolin-1 expression. Increased vascular endothelial ADAM17 activity and soluble TNF release in AT |
| Candela et al. 2017 [58] Arterioscler Thromb Vasc | Mice model | Mice obese vs. lean | Role of macrophages in determining vascular [H2S] and vasodilatation | Vascular H2S depletion sustains the loss of perivascular adipose tissue anticontractile function in obesity. |
| Xia et al. 2016 Arterioscler Thromb Vasc Bio [59] | Mice model | C57BL/6J fat diet with or without PVAT | Contribution of PVAT to vascular dysfunction. | Diet-induced obesity leads to l-arginine deficiency and eNOS uncoupling in PVAT |
| Bussey et al. 2018 Arterioscler Thromb Vasc Biol [60] | Rat Model | Mesenteric arteries with and without PVAT from control | PVAT function after weight loss induced by caloric restriction | Diet-induced weight loss reverses obesity-induced PVAT damage due to reduced inflammation and increased nitric oxide synthase activity within PVAT |
3.3. Perivascular Adipose Tissue
3.4. AMP-Activated Protein Kinase
3.5. Potassium Channels
| First Author/Year Ref | Type of Study | Cohort | Aims | Findings |
|---|---|---|---|---|
| Seki et al. 2017 J Am Heart Assoc [67] | Rat Model | SHRs with LCZ696 or valsartan Vs SHRs without LCZ696 or valsartan Vs WK | Wheter Angiotensin II receptor-neprilysin inhibitor sacubitril/valsartan. (LCZ696) would improve reduced EDH-mediated responses. | LCZ696 is effective as valsartan in improving the impaired EDH-mediated responses during hypertension. LCZ696 and valsartan exert beneficial effects on endothelium-independent relaxation by the ATP-sensitive K+ channel. |
| Jin et al. 2021 Sci Rep [71] | Rat model | HPH rats in vivo PMVEC in vitro | Whether CTRP9 has protective roles in the development of HPH. | CTRP9 lead to NO production and reduces ET-1 production by regulating AMPK activation. CTRP9 could be a target for HPH. |
| Omura et al. 2016 Circ Res. 2016 [75] | Mice model | eAMPK (−/−) eAMPK(flox/flox) | To determine the role of endothelial AMPK in the development of PAH. | Novel therapeutic target for the treatment of PAH exerted by endothelial AMPK. |
| Shentu et al. 2016 Arterioscler Thromb Vasc Biol [80] | Human model Mice model | Human umbilical vein ECs EC-specific AMPKα2 | Whether AMPK and SIRT1 coregulate cortactin dynamics in response to shear stress. | AMPK/SIRT1 coregulated cortactin-F-actin dynamics is required for endothelial nitric oxide synthase. Atheroprotective role of AMPK/SIRT1. |
| Li et al. 2016 Arterioscler Thromb Vasc [82] | Mice model | db/db mice Infusion miR-34a inhibitor Genetic ablation of endothelial miR-34a | Role of endothelial miR-34a in diabetic vascular dysfunction by targeting Sirt1. | Endothelial upregulation of miR-34a leads to endothelial dysfunction by targeting Sirt1. |
| Stott et al. 2018 [85] Arterioscler Thromb Vasc | Rat model | RA vs. MA | The role of Kv7 channels in EPAC dependent relaxations of the rat vasculature. | EPAC-dependent vasorelaxations occur in part via activation of Kv7 channels. Effect in mesenteric, but not renal arteries. |
| Lindman et al. 2018 Ipertension [87] | In vitro rat model | WK RA Vs WK MA | The role of microtubule stability on β-adrenoceptor signaling in rat renal and mesenteric arteries. | Microtubule disruption improves the β-adrenoceptor-mediated relaxations of mesenteric and renal arteries. |
| Xu et al. 2016 [91] Arterioscler Thromb Vasc | Mice model | Apolipoprotein E knockout mice LRA Vs Apolipoprotein E knockout mice LCCA | Role of Kca3.1 I in macrophage polarization. Relationship to plaque instability. | Block of kCa3.1 suppresses plaque instability. |
| Hu et al. 2016 [91] Arterioscler Thromb Vasc | Mice model | Ad-Bmp4 Ad-Pdgfa-shRNA | Whether PDGF mediates BMP4-induced endothelial dysfunction in diabetes mellitus. | PDGF-AA impairs endothelium-dependent vasodilation. PDGF-AA mediates BMP4-induced adverse effect on endothelial cell function through SMAD1/5- and SMAD4-dependent mechanisms. Inhibition of PGDF-AA ameliorates vascular dysfunction. |
| Chen 2017 [92] Arterioscler Thromb Vasc | Mice model | EC-specific P2Y2R-deficient mice EC-specific P2Y2R knockout mice onto an ApoE−/− | Role of endothelial P2Y2R in the pathogenesis of atherosclerosis. | EC-specific P2Y2R deficiency reduces atherosclerotic burden and promotes plaque stability in ApoE−/− mice. |
3.6. Bone Morphogenic Protein 4
3.7. P2Y2 Receptor
4. Endothelin and Cardiovascular Disease
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CABG | coronary artery bypass grafting |
| CAD | coronary artery disease |
| cGMP | cyclic guanosine monophosphate |
| EDH | endothelium-dependent hyperpolarization |
| EDRF | endothelium-derived relaxing factor |
| EC | Endothelial cell |
| H2O2 | hydrogen peroxide |
| LITA | left internal thoracic mammary |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NO | nitric oxide |
| P2Y2 | purinoceptor 2 |
| PVAT | Perivascular Adipose Tissue |
| RCT | randomized clinical trial |
References
- Luksha, L.; Agewall, S.; Kublickiene, K. Endothelium-derived hyperpolarizing factor in vascular physiology and cardiovascular disease. Atherosclerosis 2009, 202, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, H. 2014 Williams Harvey Lecture: Importance of coronary vasomotion abnormalities—from bench to bedside. Eur. Heart J. 2014, 35, 3180–3193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohashi, J.; Sawada, A.; Nakajima, S.; Noda, K.; Takaki, A.; Shimokawa, H. Mechanisms for enhanced endothelium-derived hy-perpolarizing factor-mediated responses in microvessels in mice. Circ. J. 2012, 76, 1768–1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimokawa, H. Cellular and Molecular Mechanisms of Coronary Artery Spasm: Lessons from animal models. Jpn. Circ. J. 2000, 64, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhoutte, P.M.; Shimokawa, H.; Feletou, M.; Tang, E.H.C. Endothelial dysfunction and vascular disease—A 30th anniversary update. Acta Physiol. 2015, 219, 22–96. [Google Scholar] [CrossRef]
- Vanhoutte, P.M.; Shimokawa, H.; Tang, E.H.; Feletou, M. Endothelial dysfunction and vascular disease. Acta Physiol. 2009, 196, 193–222. [Google Scholar] [CrossRef] [Green Version]
- Vanhoutte, P.M. Endothelial Dysfunction the First Step toward Coronary Arteriosclerosis. Circ. J. 2009, 73, 595–601. [Google Scholar] [CrossRef] [Green Version]
- Loader, J.; Meziat, C.; Watts, R.; Lorenzen, C.; Sigaudo-Roussel, D.; Stewart, S.; Reboul, C.; Meyer, G.; Walther, G. Effects of Sugar-Sweetened Beverage Consumption on Microvascular and Macrovascular Function in a Healthy Population. Arter. Thromb. Vasc. Biol. 2017, 37, 1250–1260. [Google Scholar] [CrossRef] [Green Version]
- Loader, J.; Montero, D.; Lorenzen, C.; Watts, R.; Méziat, C.; Reboul, C.; Stewart, S.; Walther, G. Acute hyperglycemia impairs vascular function in healthy and cardiometabolic diseased subjects: Systematic review and meta-analysis. Arter. Thromb. Vasc. Biol. 2015, 35, 2060–2072. [Google Scholar] [CrossRef] [Green Version]
- Bretón-Romero, R.; Feng, B.; Holbrook, M.; Farb, M.G.; Fetterman, J.L.; Linder, E.A.; Berk, B.D.; Masaki, N.; Weisbrod, R.M.; Inagaki, E.; et al. Endothelial Dysfunction in Human Diabetes Is Mediated by Wnt5a–JNK Signaling. Arter. Thromb. Vasc. Biol. 2016, 36, 561–569. [Google Scholar] [CrossRef]
- Walther, G.; Obert, P.; Dutheil, F.; Chapier, R.; Lesourd, B.; Naughton, G.; Courteix, D.; Vinet, A. Metabolic Syndrome Individuals with and without Type 2 Diabetes Mellitus Present Generalized Vascular Dysfunction. Arter. Thromb. Vasc. Biol. 2015, 35, 1022–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manríquez-Núñez, J.; Ramos-Gómez, M. Bioactive Compounds and Adipocyte Browning Phenomenon. Curr. Issues Mol. Biol. 2022, 44, 3039–3052. [Google Scholar] [CrossRef] [PubMed]
- Hazra, S.; Henson, G.D.; Bramwell, R.C.; Donato, A.J.; Lesniewski, L.A. Impact of high-fat diet on vasoconstrictor reactivity of white and brown adipose tissue resistance arteries. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H485–H494. [Google Scholar] [CrossRef] [PubMed]
- Nowak, W.; Deng, J.; Ruan, X.Z.; Xu, Q. Reactive Oxygen Species Generation and Atherosclerosis. Arter. Thromb. Vasc. Biol. 2017, 37, e41–e52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonetti, P.O.; Pumper, G.M.; Higano, S.T.; Holmes, D.R., Jr.; Kuvin, J.T.; Lerman, A. Noninvasive identification of patients with early coronary atherosclerosis by assessment of digital reactive hyperemia. J. Am. Coll. Cardiol. 2004, 44, 2137–2141. [Google Scholar] [CrossRef] [Green Version]
- Kitta, Y.; Obata, J.-E.; Nakamura, T.; Hirano, M.; Kodama, Y.; Fujioka, D.; Saito, Y.; Kawabata, K.-I.; Sano, K.; Kobayashi, T.; et al. Persistent Impairment of Endothelial Vasomotor Function Has a Negative Impact on Outcome in Patients with Coronary Artery Disease. J. Am. Coll. Cardiol. 2009, 53, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Matsuzawa, Y.; Sugiyama, S.; Sugamura, K.; Nozaki, T.; Ohba, K.; Konishi, M.; Matsubara, J.; Sumida, H.; Kaikita, K.; Kojima, S.; et al. Digital Assessment of Endothelial Function and Ischemic Heart Disease in Women. J. Am. Coll. Cardiol. 2010, 55, 1688–1696. [Google Scholar] [CrossRef] [Green Version]
- Matsuzawa, Y.; Kwon, T.; Lennon, R.J.; Lerman, L.O.; Lerman, A. Prognostic Value of Flow-Mediated Vasodilation in Brachial Artery and Fingertip Artery for Cardiovascular Events: A Systematic Review and Meta-Analysis. J. Am. Heart Assoc. 2015, 4, e002270. [Google Scholar] [CrossRef] [Green Version]
- Takase, S.; Matoba, T.; Nakashiro, S.; Mukai, Y.; Inoue, S.; Oi, K.; Higo, T.; Katsuki, S.; Takemoto, M.; Suematsu, N.; et al. Ezetimibe in Combination with Statins Ameliorates Endothelial Dysfunction in Coronary Arteries after stenting: The CuVIC trial (effect of cholesterol absorption inhibitor usage on target vessel dysfunction after cor-onary stenting), a multicenter randomized controlled trial. Arter. Thromb. Vasc. Biol. 2017, 37, 350–358. [Google Scholar] [CrossRef] [Green Version]
- Cannon, C.P.; Blazing, M.A.; Giugliano, R.P.; McCagg, A.; White, J.A.; Théroux, P.; Darius, H.; Lewis, B.S.; Ophuis, T.O.; Jukema, J.W.; et al. Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. N. Engl. J. Med. 2015, 372, 2387–2397. [Google Scholar] [CrossRef]
- Lin, X.; Racette, S.B.; Ma, L.; Wallendorf, M.; Ostlund, J.R.E. Ezetimibe Increases Endogenous Cholesterol Excretion in Humans. Arter. Thromb. Vasc. Biol. 2017, 37, 990–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Xiong, S.; Lin, S.; Xia, W.; Li, Q.; Zhao, Z.; Wei, X.; Lu, Z.; Wei, X.; Gao, P.; et al. Enhanced Mitochondrial Transient Receptor Potential Channel, Canonical Type 3-Mediated Calcium Handling in the Vasculature From Hypertensive Rats. Am. Heart Assoc. 2017, 6, e005812. [Google Scholar] [CrossRef] [PubMed]
- Montezano, A.C.; De Lucca Camargo, L.; Persson, P.; Rios, F.J.; Harvey, A.P.; Anagnostopoulou, A.; Palacios, R.; Gandara, A.C.P.; Alves-Lopes, R.; Neves, K.B.; et al. RMNADPH Oxidase 5 Is a Pro-Contractile Nox Isoform and a Point of Cross-Talk for Calcium and Redox Signaling-Implications in Vascular Function. J. Am. Heart Assoc. 2018, 7, e009388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikumi, Y.I.; Shiroto, T.; Godo, S.; Saito, H.; Tanaka, S.; Ito, A.; Kajitani, S.; Monma, Y.; Miyata, S.; Tsutsui, M.; et al. Important Roles of Endothelium-Dependent Hyperpolarization in Coronary Microcirculation and Cardiac Diastolic Function in Mice. J. Cardiovasc. Pharmacol. 2020, 75, 31–40. [Google Scholar] [CrossRef]
- Hartigh, L.J.D.; Omer, M.; Goodspeed, L.; Wang, S.; Wietecha, T.; O’Brien, K.D.; Han, C.Y. Adipocyte-Specific Deficiency of NADPH Oxidase 4 Delays the Onset of Insulin Resistance and Attenuates Adipose Tissue Inflammation in Obesity. Arter. Thromb. Vasc. Biol. 2017, 37, 466–475. [Google Scholar] [CrossRef] [Green Version]
- Sano, T.; Nagayasu, S.; Suzuki, S.; Iwashita, M.; Yamashita, A.; Shinjo, T.; Sanui, T.; Kushiyama, A.; Kanematsu, T.; Nishimura, F. Epicatechin downregulates adipose tissue CCL19 expression and thereby ameliorates diet-induced obesity and insulin resistance. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 249–259. [Google Scholar] [CrossRef]
- Li, A.; Shi, W.; Wang, J.; Wang, X.; Zhang, Y.; Lei, Z.; Jiao, X.-Y. The gene knockout of angiotensin II type 1a receptor improves high-fat diet-induced obesity in rat via promoting adipose lipolysis. PLoS ONE 2022, 17, e0267331. [Google Scholar] [CrossRef]
- La Favor, J.D.; Dubis, G.S.; Yan, H.; White, J.D.; Nelson, M.A.; Anderson, E.J.; Hickner, R.C. Microvascular Endothelial Dysfunction in Sedentary, Obese Humans Is Mediated by NADPH Oxidase: Influence of exercise training. Arter. Thromb. Vasc. Biol. 2016, 36, 2412–2420. [Google Scholar] [CrossRef] [Green Version]
- Masi, S.; Colucci, R.; Duranti, E.; Nannipieri, M.; Anselmino, M.; Ippolito, C.; Tirotta, E.; Georgiopoulos, G.; Garelli, F.; Nericcio, A.; et al. Aging Modulates the Influence of Arginase on Endothelial Dysfunction in Obesity. Arter. Thromb. Vasc. Biol. 2018, 38, 2474–2483. [Google Scholar] [CrossRef] [Green Version]
- Godo, S.; Sawada, A.; Saito, H.; Ikeda, S.; Enkhjargal, B.; Suzuki, K.; Tanaka, S.; Shimokawa, H. Disruption of Physiological Balance Between Nitric Oxide and Endothelium-Dependent Hyperpolarization Impairs Cardiovascular Homeostasis in Mice. Arter. Thromb. Vasc. Biol. 2016, 36, 97–107. [Google Scholar] [CrossRef]
- Gray, S.P.; Di Marco, E.; Kennedy, K.; Chew, P.; Okabe, J.; El-Osta, A.; Calkin, A.C.; Biessen, E.A.; Touyz, R.M.; Cooper, M.E.; et al. Reactive Oxygen Species Can Provide Atheroprotection via NOX4-Dependent Inhibition of Inflammation and Vascular Remodeling. Arter. Thromb. Vasc. Biol. 2016, 36, 295–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otsuka, F.; Yahagi, K.; Sakakura, K.; Virmani, R. Why is the mammary artery so special and what protects it from atherosclerosis? Ann. Cardiothorac. Surg. 2013, 2, 519–526. [Google Scholar] [PubMed]
- Freed, J.K.; Beyer, A.M.; Logiudice, J.A.; Hockenberry, J.C.; Gutterman, D.D. Ceramide Changes the Mediator of Flow-Induced Vasodilation From Nitric Oxide to Hydrogen Peroxide in the Human Microcirculation. Circ. Res. 2014, 115, 525–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durand, M.J.; Zinkevich, N.S.; Riedel, M.; Gutterman, D.D.; Nasci, V.L.; Salato, V.K.; Hijjawi, J.B.; Reuben, C.F.; North, P.E.; Beyer, A.M. Vascular actions of angiotensin 1-7 in the human microcirculation: Novel role for telomerase. Arter. Thromb. Vasc. Biol. 2016, 36, 1254–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirsch, J.; Schneider, H.; Pagel, J.-I.; Rehberg, M.; Singer, M.; Hellfritsch, J.; Chillo, O.; Schubert, K.M.; Qiu, J.; Pogoda, K.; et al. Endothelial Dysfunction, and A Prothrombotic, Proinflammatory Phenotype Is Caused by Loss of Mitochondrial Thioredoxin Reductase in Endothelium. Arter. Thromb. Vasc. Biol. 2016, 36, 1891–1899. [Google Scholar] [CrossRef] [Green Version]
- Erbs, S.; Möbius-Winkler, S.; Linke, A.; Adams, V.; Doll, N.; Gielen, S.; Gummert, J.F.; Mohr, F.W.; Schuler, G.; Hambrecht, R. Both T-786C and G894T polymorphism of endothelial nitric oxide synthase affect in-vitro endothelium-dependent relaxation of internal mammary artery rings from patients with coronary artery disease. Eur. J. Cardiovasc. Prev. Rehabil. 2006, 13, 826–831. [Google Scholar] [CrossRef]
- Lee, M.Y.; Griendling, K.K. Redox signaling, vascular function, and hypertension. Antioxid. Redox. Signal. 2008, 10, 1045–1059. [Google Scholar] [CrossRef]
- Godo, S.; Shimokawa, H. Divergent roles of endothelial nitric oxide synthases system in maintaining cardiovascular homeostasis. Free Radic. Biol. Med. 2017, 109, 4–10. [Google Scholar] [CrossRef]
- Shimokawa, H.; Godo, S. Nitric oxide and endothelium-dependent hyperpolarization mediated by hydrogen peroxide in health and disease. Basic Clin. Pharmacol. Toxicol. 2020, 127, 92–101. [Google Scholar] [CrossRef]
- Han, C.Y. Roles of Reactive Oxygen Species on Insulin Resistance in Adipose Tissue. Diabetes Metab. J. 2016, 40, 272–279. [Google Scholar] [CrossRef]
- Sano, T.; Iwashita, M.; Nagayasu, S.; Yamashita, A.; Shinjo, T.; Hashikata, A.; Asano, T.; Kushiyama, A.; Ishimaru, N.; Takahama, Y.; et al. Protection from diet-induced obesity and insulin resistance in mice lacking CCL19-CCR7 signaling. Obesity 2015, 23, 1460–1471. [Google Scholar] [CrossRef] [PubMed]
- Gutterman, D.D.; Chabowski, D.S.; Kadlec, A.O.; Durand, M.J.; Freed, J.K.; Ait-Aissa, K.; Beyer, A.M. The human microcirculation: Reg-ulation of flow and beyond. Circ. Res. 2016, 118, 157–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmström, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Kadlec, A.O.; Chabowski, D.S.; Ait-Aissa, K.; Gutterman, D.D. Role of PGC-1α in Vascular Regulation: Implications for atheroscle-rosis. Arter. Thromb. Vasc. Biol. 2016, 36, 1467–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erbs, S.; Baither, Y.; Linke, A.; Adams, V.; Shu, Y.; Lenk, K.; Gielen, S.; Dilz, R.; Schuler, G.; Hambrecht, R. Promoter but Not Exon 7 Polymorphism of Endothelial Nitric Oxide Synthase Affects Training-Induced Correction of Endothelial Dysfunction. Arter. Thromb. Vasc. Biol. 2003, 23, 1814–1819. [Google Scholar] [CrossRef] [PubMed]
- Cattaruzza, M.; Guzik, T.J.; Słodowski, W.; Pelvan, A.; Becker, J.; Halle, M.; Buchwald, A.B.; Channon, K.M.; Hecker, M. Shear Stress Insensitivity of Endothelial Nitric Oxide Synthase Expression as a Genetic Risk Factor for Coronary Heart Disease. Circ. Res. 2004, 95, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Lüscher, T.F.; Corti, R. Flow: The signal of life. Circ. Res. 2004, 95, 749–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, J.-I.; Berk, B.C. Novel Mechanisms of Endothelial Mechanotransduction. Arter. Thromb. Vasc. Biol. 2014, 34, 2378–2386. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Li, Y.-S.; Chien, S. Shear Stress–Initiated Signaling and Its Regulation of Endothelial Function. Arter. Thromb. Vasc. Biol. 2014, 34, 2191–2198. [Google Scholar] [CrossRef] [Green Version]
- Warboys, C.M.; de Luca, A.; Amini, N.; Luong, L.; Duckles, H.; Hsiao, S.; White, A.; Biswas, S.; Khamis, R.; Chong, C.K.; et al. Disturbed Flow Promotes Endothelial Senescence via a p53-Dependent Pathway. Arter. Thromb. Vasc. Biol. 2014, 34, 985–995. [Google Scholar] [CrossRef]
- Shindo, T.; Ito, K.; Ogata, T.; Hatanaka, K.; Kurosawa, R.; Eguchi, K.; Kagaya, Y.; Hanawa, K.; Aizawa, K.; Shiroto, T.; et al. Low-Intensity Pulsed Ultrasound Enhances Angiogenesis and Ameliorates Left Ventricular Dysfunction in a Mouse Model of Acute Myocardial Infarction. Arter. Thromb. Vasc. Biol. 2016, 36, 1220–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatanaka, K.; Ito, K.; Shindo, T.; Kagaya, Y.; Ogata, T.; Eguchi, K.; Kurosawa, R.; Shimokawa, H. Molecular mechanisms of the angiogenic effects of low-energy shock wave therapy: Roles of mechanotransduction. Am. J. Physiol. Physiol. 2016, 311, C378–C385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friederich-Persson, M.; Cat, A.N.D.; Persson, P.; Montezano, A.C.; Touyz, R.M. Brown Adipose Tissue Regulates Small Artery Function Through NADPH Oxidase 4–Derived Hydrogen Peroxide and Redox-Sensitive Protein Kinase G-1α. Arter. Thromb. Vasc. Biol. 2017, 37, 455–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgoyne, J.R.; Madhani, M.; Cuello, F.; Charles, R.L.; Brennan, J.P.; Schröder, E.; Browning, D.D.; Eaton, P. Cysteine redox sensor in PKGIa enables oxidant-induced activation. Science 2007, 317, 1393–1397. [Google Scholar] [CrossRef] [PubMed]
- Prysyazhna, O.; Rudyk, O.; Eaton, P. Single atom substitution in mouse protein kinase G eliminates oxidant sensing to cause hypertension. Nat. Med. 2012, 18, 286–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noblet, J.N.; Owen, M.K.; Goodwill, A.G.; Sassoon, D.J.; Tune, J.D. Lean and Obese Coronary Perivascular Adipose Tissue Impairs Vasodilation via Differential Inhibition of Vascular Smooth Muscle K+ Channels. Arter. Thromb. Vasc. Biol. 2015, 35, 1393–1400. [Google Scholar] [CrossRef] [Green Version]
- Dou, H.; Feher, A.; Davila, A.C.; Romero, M.J.; Patel, V.S.; Kamath, V.M.; Gooz, M.B.; Rudic, R.D.; Lucas, R.; Fulton, D.J.; et al. Role of Adipose Tissue Endothelial ADAM17 in Age-Related Coronary Microvascular Dysfunction. Arter. Thromb. Vasc. Biol. 2017, 37, 1180–1193. [Google Scholar] [CrossRef] [Green Version]
- Candela, J.; Wang, R.; White, C. Microvascular endothelial dysfunction in obesity is driven by macrophage-dependent hydrogen sulphide depletion. Arter. Thromb. Vasc. Biol. 2017, 37, 889–899. [Google Scholar] [CrossRef] [Green Version]
- Xia, N.; Horke, S.; Habermeier, A.; Closs, E.I.; Reifenberg, G.; Gericke, A.; Mikhed, Y.; Münzel, T.; Daiber, A.; Förstermann, U.; et al. Un-coupling of endothelial nitric oxide synthase in perivascular adipose tissue of diet-induced obese mice. Arter. Thromb. Vasc. Biol. 2016, 36, 78–85. [Google Scholar] [CrossRef] [Green Version]
- Bussey, C.E.; Withers, S.B.; Aldous, R.G.; Edwards, G.; Heagerty, A.M. Obesity-Related Perivascular Adipose Tissue Damage Is Reversed by Sustained Weight Loss in the Rat. Arter. Thromb. Vasc. Biol. 2016, 36, 1377–1385. [Google Scholar] [CrossRef]
- Nedergaard, J.; Bengtsson, T.; Cannon, B. Unexpected evidence for active brown adipose tissue in adult humans. Am. J. Physiol. Metab. 2007, 293, E444–E452. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.K.; Zhou, Z.; Zhang, J.; Zeng, R.; Wu, J.; Eitzman, D.T.; Chen, Y.E.; Chang, L. Perivascular Adipose Tissue in Vascular Function and Disease: A review of current research and animal models. Arter. Thromb. Vasc. Biol. 2014, 34, 1621–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, X.-Y.; Qu, S.-L.; Xiong, W.-H.; Rom, O.; Chang, L.; Jiang, Z.-S.; Qi, X.-Y.; Qu, S.-L.; Xiong, W.-H.; Rom, O.; et al. Perivascular adipose tissue (PVAT) in atherosclerosis: A double-edged sword. Cardiovasc. Diabetol. 2018, 17, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Garcia-Barrio, M.; Jiang, Z.-S.; Chen, Y.E.; Chang, L. Roles of Perivascular Adipose Tissue in Hypertension and Atherosclerosis. Antioxid. Redox Signal. 2021, 34, 736–749. [Google Scholar] [CrossRef]
- Man, A.W.C.; Zhou, Y.; Xia, N.; Li, H. Endothelial Nitric Oxide Synthase in the Perivascular Adipose Tissue. Biomedicines 2022, 10, 1754. [Google Scholar] [CrossRef]
- Kane, M.O.; Sene, M.; Anselm, E.; Dal, S.; Schini-Kerth, V.B.; Augier, C. Role of AMP-activated Protein Kinase in NO− and EDHF-mediated Endothelium-dependent Relaxations to Red Wine Polyphenols. Indian J. Physiol. Pharmacol. 2015, 59, 369–379. [Google Scholar]
- Seki, T.; Goto, K.; Kansui, Y.; Ohtsubo, T.; Matsumura, K.; Kitazono, T. Angiotensin II Receptor–Neprilysin Inhibitor Sacubitril/Valsartan Improves Endothelial Dysfunction in Spontaneously Hypertensive Rats. J. Am. Heart Assoc. 2017, 6, e006617. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Vanhoutte, P.M.; Leung, S.W.S. Vascular adenosine monophosphate-activated protein kinase: Enhancer, brake or both? Basic Clin. Pharmacol. Toxicol. 2020, 127, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Goto, K.; Ohtsubo, T.; Kitazono, T. Endothelium-Dependent Hyperpolarization (EDH) in Hypertension: The Role of Endothelial Ion Channels. Int. J. Mol. Sci. 2018, 19, 315. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Song, P.; Zou, M.-H. AMPK and Pulmonary Hypertension: Crossroads Between Vasoconstriction and Vascular Remodeling. Front. Cell Dev. Biol. 2021, 9, 691585. [Google Scholar] [CrossRef]
- Jin, Q.; Su, H.; Yang, R.; Tan, Y.; Li, B.; Yi, W.; Dong, Q.; Zhang, H.; Xing, W.; Sun, X. C1q/TNF-related protein-9 ameliorates hypoxia-induced pulmonary hypertension by regulating secretion of endothelin-1 and nitric oxide mediated by AMPK in rats. Sci. Rep. 2021, 11, 11372. [Google Scholar] [CrossRef] [PubMed]
- Enkhjargal, B.; Godo, S.; Sawada, A.; Suvd, N.; Saito, H.; Noda, K.; Satoh, K.; Shimokawa, H. Endothelial AMP-Activated Protein Kinase Regulates Blood Pressure and Coronary Flow Responses through Hyperpolarization Mechanism in Mice. Arter. Thromb. Vasc. Biol. 2014, 34, 1505–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvatore, T.; Pafundi, P.C.; Galiero, R.; Rinaldi, L.; Caturano, A.; Vetrano, E.; Aprea, C.; Albanese, G.; Di Martino, A.; Ricozzi, C.; et al. Can Metformin Exert as an Active Drug on Endothelial Dysfunction in Diabetic Subjects? Biomedicines 2020, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Cheang, W.S.; Tian, X.Y.; Wong, W.T.; Lau, C.W.; Lee, S.S.-T.; Chen, Z.Y.; Yao, X.; Wang, N.; Huang, Y. Metformin Protects Endothelial Function in Diet-Induced Obese Mice by Inhibition of Endoplasmic Reticulum Stress through 5′ Adenosine Monophosphate–Activated Protein Kinase–Peroxisome Proliferator–Activated Receptor δ Pathway. Arter. Thromb. Vasc. Biol. 2014, 34, 830–836. [Google Scholar] [CrossRef] [Green Version]
- Omura, J.; Satoh, K.; Kikuchi, N.; Satoh, T.; Kurosawa, R.; Nogi, M.; Otsuki, T.; Kozu, K.; Numano, K.; Suzuki, K.; et al. Protective Roles of Endothelial AMP-Activated Protein Kinase against Hypoxia-Induced Pulmonary Hypertension in Mice. Circ. Res. 2016, 119, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Bai, B.; Vanhoutte, P.M.; Wang, Y. Loss-of-SIRT1 function during vascular ageing: Hyperphosphorylation mediated by cyclin-dependent kinase 5. Trends Cardiovasc. Med. 2014, 24, 81–84. [Google Scholar] [CrossRef]
- Bai, B.; Liang, Y.; Xu, C.; Lee, M.Y.; Xu, A.; Wu, N.; Vanhoutte, P.M.; Wang, Y. Cyclin-Dependent Kinase 5–Mediated Hyperphosphorylation of Sirtuin-1 Contributes to the Development of Endothelial Senescence and Atherosclerosis. Circulation 2012, 126, 729–740. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Xu, A.; Wang, Y. SIRT1 in Endothelial Cells as a Novel Target for the Prevention of Early Vascular Aging. J. Cardiovasc. Pharmacol. 2016, 67, 465–473. [Google Scholar] [CrossRef]
- Zu, Y.; Liu, L.; Lee, M.Y.; Xu, C.; Liang, Y.; Man, R.Y.; Vanhoutte, P.M.; Wang, Y. SIRT1 Promotes Proliferation and Prevents Senescence through Targeting LKB1 in Primary Porcine Aortic Endothelial Cells. Circ. Res. 2010, 106, 1384–1393. [Google Scholar] [CrossRef] [Green Version]
- Shentu, T.-P.; He, M.; Sun, X.; Zhang, J.; Zhang, F.; Gongol, B.; Marin, T.L.; Zhang, J.; Wen, L.; Wang, Y.; et al. AMP-Activated Protein Kinase and Sirtuin 1 Coregulation of Cortactin Contributes to Endothelial Function. Arter. Thromb. Vasc. Biol. 2016, 36, 2358–2368. [Google Scholar] [CrossRef] [Green Version]
- Belvitch, P.; Rizzo, A.N.; Dudek, S.M. Cortactin in atherosclerosis: Just say NO. Arter. Thromb. Vasc. Biol. 2016, 36, 2278–2280. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Kim, Y.-R.; Vikram, A.; Kumar, S.; Kassan, M.; Gabani, M.; Lee, S.K.; Jacobs, J.S.; Irani, K. P66Shc-Induced MicroRNA-34a Causes Diabetic Endothelial Dysfunction by Downregulating Sirtuin1. Arter. Thromb. Vasc. Biol. 2016, 36, 2394–2403. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Li, X.; Sha, X.; Xi, H.; Li, Y.-F.; Shao, Y.; Mai, J.; Virtue, A.; Lopez-Pastrana, J.; Meng, S.; et al. Early Hyperlipidemia Promotes Endothelial Activation via a Caspase-1-Sirtuin 1 Pathway. Arter. Thromb. Vasc. Biol. 2015, 35, 804–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, S.W.S.; Vanhoutte, P.M. Endothelium-dependent hyperpolarization: Age, gender and blood pressure, do they matter? Acta Physiol. 2015, 219, 108–123. [Google Scholar] [CrossRef] [PubMed]
- Stott, J.B.; Barrese, V.; Greenwood, I.A. Kv7 Channel Activation Underpins EPAC-Dependent Relaxations of Rat Arteries. Arter. Thromb. Vasc. Biol. 2016, 36, 2404–2411. [Google Scholar] [CrossRef] [Green Version]
- Stott, J.B.; Barrese, V.; Suresh, M.; Masoodi, S.; Greenwood, I.A. Investigating the Role of G Protein βγ in Kv7-Dependent Relaxations of the Rat Vasculature. Arter. Thromb. Vasc. Biol. 2018, 38, 2091–2102. [Google Scholar] [CrossRef] [Green Version]
- Lindman, J.; Khammy, M.M.; Lundegaard, P.R.; Aalkjær, C.; Jepps, T.A. Microtubule Regulation of Kv7 Channels Orchestrates cAMP-Mediated Vasorelaxations in Rat Arterial Smooth Muscle. Hypertension 2018, 71, 336–345. [Google Scholar] [CrossRef]
- Xu, R.; Li, C.; Wu, Y.; Shen, L.; Ma, J.; Qian, J.; Ge, J. Role of KCa3.1 channels in macrophage polarization and its relevance in ather-osclerotic plaque instability. Arter. Thromb. Vasc. Biol. 2017, 37, 226–236. [Google Scholar] [CrossRef] [Green Version]
- Köhler, R.; Ruth, P. Endothelial dysfunction and blood pressure alterations in K+-channel transgenic mice. Pflug. Arch. 2010, 459, 969–976. [Google Scholar] [CrossRef]
- Wulff, H.; Köhler, R. Endothelial Small-Conductance and Intermediate-Conductance KCa Channels: An update on their phar-macology and usefulness as cardiovascular targets. J. Cardiovasc. Pharmacol. 2013, 61, 102–112. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Zhang, Y.; Wang, L.; Lau, C.W.; Xu, J.; Luo, J.-Y.; Gou, L.; Yao, X.; Chen, Z.-Y.; Ma, R.C.W.; et al. Bone Morphogenic Protein 4-Smad–Induced Upregulation of Platelet-Derived Growth Factor AA Impairs Endothelial Function. Arter. Thromb. Vasc. Biol. 2016, 36, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Qian, S.; Hoggatt, A.; Tang, H.; Hacker, T.A.; Obukhov, A.G.; Herring, P.B.; Seye, C.I. Endothelial Cell–Specific Deletion of P2Y2 Receptor Promotes Plaque Stability in Atherosclerosis-Susceptible ApoE-Null Mice. Arter. Thromb. Vasc. Biol. 2017, 37, 75–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, W.T.; Tian, X.Y.; Chen, Y.; Leung, F.P.; Liu, L.; Lee, H.K.; Ng, C.-F.; Xu, A.; Yao, X.; Vanhoutte, P.M.; et al. Bone Morphogenic Protein-4 Impairs Endothelial Function Through Oxidative Stress–Dependent Cyclooxygenase-2 Upregulation: Implica-tions on hypertension. Circ. Res. 2010, 107, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.M.; Zhang, Y.; Huang, Y. Bone morphogenic protein-4-induced oxidant signaling via protein carbonylation for endothelial dysfunction. Free Radic. Biol. Med. 2014, 75, 178–190. [Google Scholar] [CrossRef]
- Shen, S.; Wang, F.; Fernandez, A.; Hu, W. Role of platelet-derived growth factor in type II diabetes mellitus and its complications. Diabetes Vasc. Dis. Res. 2020, 17, 1479164120942119. [Google Scholar] [CrossRef]
- Lau, Y.S.; Tian, X.Y.; Mustafa, M.R.; Murugan, D.; Liu, J.; Zhang, Y.; Lau, C.W.; Huang, Y. Boldine improves endothelial function in diabetic db/db mice through inhibition of angiotensin II-mediated BMP4-oxidative stress cascade. Br. J. Pharmacol. 2013, 170, 1190–1198. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, J.; Tian, X.Y.; Wong, W.T.; Chen, Y.; Wang, L.; Luo, J.; Cheang, W.S.; Lau, C.W.; Kwan, K.M.; et al. Inhibition of Bone Morphogenic Protein 4 Restores Endothelial Function in db/db Diabetic Mice. Arter. Thromb. Vasc. Biol. 2014, 34, 152–159. [Google Scholar] [CrossRef] [Green Version]
- Qian, S.; Regan, J.N.; Shelton, M.T.; Hoggatt, A.; Mohammad, K.S.; Herring, P.B.; Seye, C.I. The P2Y 2 nucleotide receptor is an inhibitor of vascular calcification. Atherosclerosis 2017, 257, 38–46. [Google Scholar] [CrossRef] [Green Version]
- Schwanekamp, J.A.; Lorts, A.; Vagnozzi, R.J.; Vanhoutte, D.; Molkentin, J.D. Deletion of Periostin Protects Against Atherosclerosis in Mice by Altering Inflammation and Extracellular Matrix Remodeling. Arter. Thromb. Vasc. Biol. 2016, 36, 60–68. [Google Scholar] [CrossRef] [Green Version]
- Bachschmid, M.M.; Schildknecht, S.; Matsui, R.; Zee, R.; Haeussler, D.; Cohen, R.A.; Pimental, D.; van der Loo, B. Vascular aging: Chronic oxidative stress and impairment of redox signaling—Consequences for vascular homeostasis and disease. Ann. Med. 2011, 45, 17–36. [Google Scholar] [CrossRef] [Green Version]
- Cockcroft, J.; Mancia, G. Vascular aging: Shifting the paradigm of risk assessment and reduction in hypertension. J. Hypertens. 2012, 30, S1–S2. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, M.; Kurihara, H.; Kimura, S.; Tomobe, Y.; Kobayashi, M.; Mitsui, Y.; Yazaki, Y.; Goto, K.; Masaki, T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 1988, 332, 411–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davenport, A.P.; Hyndman, K.A.; Dhaun, N.; Southan, C.; Kohan, D.E.; Pollock, J.S.; Pollock, D.M.; Webb, D.J.; Maguire, J.J. Endothelin. Pharmacol. Rev. 2016, 68, 357–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardillo, C.; Kilcoyne, C.M.; Waclawiw, M.; Cannon, I.R.O.; Panza, J.A. Role of Endothelin in the Increased Vascular Tone of Patients with Essential Hypertension. Hypertension 1999, 33, 753–758. [Google Scholar] [CrossRef] [Green Version]
- Cau, S.B.D.A.; Carneiro, F.S.; Tostes, R.C. Differential Modulation of Nitric Oxide Synthases in Aging: Therapeutic Opportunities. Front. Physiol. 2012, 3, 218. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Barber, D.A.; Lewis, D.A.; McGregor, C.G.A.; Sieck, G.C.; Fitzpatrick, L.A.; Miller, V.M. Gender and transcriptional regulation of NO synthase and ET-1 in porcine aortic endothelial cells. Am. J. Physiol. Circ. Physiol. 1997, 273, H1962–H1967. [Google Scholar] [CrossRef]
- Cramer, H.; Müller-Esterl, W.; Schroeder, C. Subtype-Specific Desensitization of Human Endothelin ETA and ETB Receptors Reflects Differential Receptor Phosphorylation. Biochemistry 1997, 36, 13325–13332. [Google Scholar] [CrossRef]
- David, F.L.; Carvalho, M.H.C.; Cobra, A.L.; Nigro, D.; Fortes, Z.B.; Rebouças, N.A.; Tostes, R.C. Ovarian Hormones Modulate Endothelin-1 Vascular Reactivity and mRNA Expression in DOCA-Salt Hypertensive Rats. Hypertension 2001, 38 Pt 2, 692–696. [Google Scholar] [CrossRef] [Green Version]
- Del Ry, S.; Maltinti, M.; Giannessi, D.; Cavallini, G.; Bergamini, E. Age-Related Changes in Endothelin-1 Receptor Subtypes in Rat Heart. Exp. Aging Res. 2008, 34, 251–266. [Google Scholar] [CrossRef]
- Zhang, C.-L.; Xie, S.; Qiao, X.; An, Y.-M.; Zhang, Y.; Li, L.; Guo, X.-B.; Zhang, F.-C.; Wu, L.-L. Plasma endothelin-1-related peptides as the prognostic biomarkers for heart failure. Medicine 2017, 96, e9342. [Google Scholar] [CrossRef]
- Yokoi, K.; Adachi, H.; Hirai, Y.; Enomoto, M.; Fukami, A.; Ogata, K.; Tsukagawa, E.; Kasahara, A.; Imaizumi, T. Plasma Endothelin-1 Level Is a Predictor of 10-Year Mortality in a General Population. Circ. J. 2012, 76, 2779–2784. [Google Scholar] [CrossRef] [PubMed]
- Kuczmarski, A.V.; Welti, L.M.; Moreau, K.L.; Wenner, M.M. ET-1 as a Sex-Specific Mechanism Impacting Age-Related Changes in Vascular Function. Front. Aging 2021, 2, 727416. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nappi, F.; Fiore, A.; Masiglat, J.; Cavuoti, T.; Romandini, M.; Nappi, P.; Avtaar Singh, S.S.; Couetil, J.-P. Endothelium-Derived Relaxing Factors and Endothelial Function: A Systematic Review. Biomedicines 2022, 10, 2884. https://doi.org/10.3390/biomedicines10112884
Nappi F, Fiore A, Masiglat J, Cavuoti T, Romandini M, Nappi P, Avtaar Singh SS, Couetil J-P. Endothelium-Derived Relaxing Factors and Endothelial Function: A Systematic Review. Biomedicines. 2022; 10(11):2884. https://doi.org/10.3390/biomedicines10112884
Chicago/Turabian StyleNappi, Francesco, Antonio Fiore, Joyce Masiglat, Teresa Cavuoti, Michela Romandini, Pierluigi Nappi, Sanjeet Singh Avtaar Singh, and Jean-Paul Couetil. 2022. "Endothelium-Derived Relaxing Factors and Endothelial Function: A Systematic Review" Biomedicines 10, no. 11: 2884. https://doi.org/10.3390/biomedicines10112884