Microbiol. Res. 2023, 14(1), 205-217; https://doi.org/10.3390/microbiolres14010017 - 6 Feb 2023
Cited by 3 | Viewed by 1962
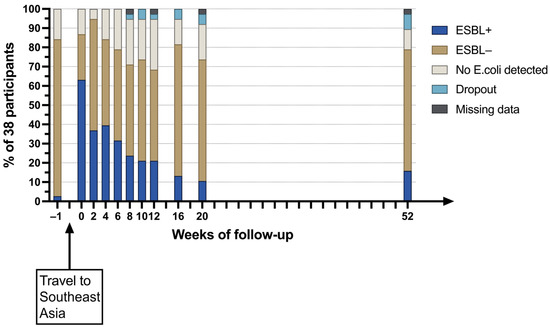
Abstract
►
Show Figures
Hops contain a wide variety of polyphenolic compounds with diverse antimicrobial properties. This study aimed to evaluate the impact of temperature on the bioactive components of samples of aqueous extracts of hops with different characteristics. A central compound rotating design model was used
[...] Read more.
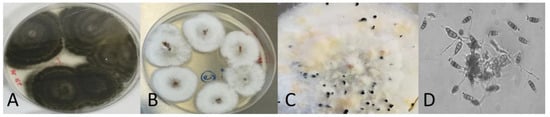
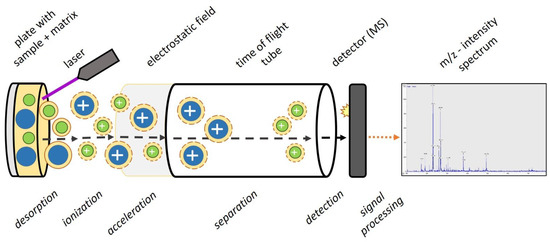
Hops contain a wide variety of polyphenolic compounds with diverse antimicrobial properties. This study aimed to evaluate the impact of temperature on the bioactive components of samples of aqueous extracts of hops with different characteristics. A central compound rotating design model was used in order to obtain optimal conditions of temperature and extract concentration to inhibit Fusarium oxysporum and Alternaria solani. At intermediate temperatures according to the design of experiments, significant effects on antifungal activity were observed. The optimal conditions with antifungal activity were at a concentration of 160 mg/mL and a temperature of 65 °C to obtain mycelial diameters ≤ 25 mm. The bioactive compounds were shown in the FT-IR spectrum after each heat treatment of both samples; significant changes were observed in the bands between 2786 to 3600 cm−1 and 1022 to 1729 cm−1. The content of total phenols and flavonoids showed a concentration increase of 4.54 to 6.24 mg GAE/g and 6.21 to 8.12 mg QE/g from an initial evaluation temperature of 25 °C to 57.5 °C, respectively, benefited by the heating temperature, enhancing antifungal activity. However, when increasing the temperature ≥90 °C, a tendency to decrease the concentration of bioactive compounds was observed, probably due to their denaturation due to the effect of temperature and exposure time, being non-thermolabile compounds at high temperatures. These aqueous extracts are an alternative to effective natural antifungals.
Full article
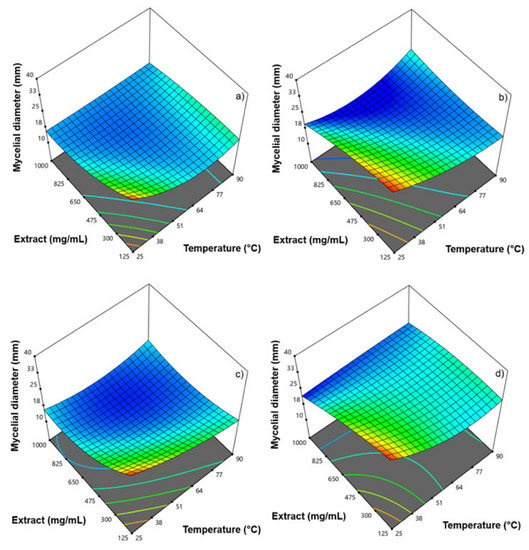
Figure 1









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}