
Abstract
Bardet–Biedl syndrome (BBS) is a model disease for ciliopathy in humans. The remarkable genetic heterogeneity that characterizes this disease is consistent with accumulating data on the interaction between the proteins encoded by the 14 BBS genes identified to date. Previous reports suggested that such interaction may also extend to instances of oligogenic inheritance in the form of triallelism which defies the long held view of BBS as an autosomal recessive disease. In order to investigate the magnitude of triallelism in BBS, we conducted a comprehensive analysis of all 14 BBS genes as well as the CCDC28B-modifier gene in a cohort of 29 BBS families, most of which are multiplex. Two in trans mutations in a BBS gene were identified in each of these families for a total of 20 mutations including 12 that are novel. In no instance did we observe two mutations in unaffected members of a given family, or observe the presence of a third allele that convincingly acted as a modifier of penetrance and supported the triallelic model of BBS. In addition to presenting a comprehensive genotype/phenotype overview of a large set of BBS mutations, including the occurrence of nonsyndromic retinitis pigmentosa in a family with a novel BBS9 mutation, our study argues in favor of straightforward autosomal recessive BBS in most cases.
Similar content being viewed by others


Introduction
Bardet–Biedl syndrome (BBS) is a multisystem disorder characterized by retinal degeneration, obesity, polydactyly, cognitive and renal impairment and hypogenitalism.1, 2, 3, 4 These and other primary and secondary features of BBS are now known to reflect the phenotypic consequences of impaired ciliary function or ciliopathy, an expansive group of developmental disorders among which BBS features prominently.5 Although much remains to be learned about the formation and function of these cellular appendages, a model has emerged in which postmitotic cells, no longer requiring centrioles to organize their mitotic spindles, migrate their centrioles close to the cell membrane where they form a basal body upon which a highly organized cytoskeletal structure starts budding until it forms a mature cilium that interacts with the surrounding environment via a repertoire of signaling cascades the integrity of which is dependent on intact antero- and retrograde intraflagellar transport.6 Despite the clear demonstration of impaired ciliary function in all genetic models of BBS, the relationship between the various BBS genes (14 described to date) and cilia is not always straightforward and involves trafficking of vesicles for intraflagellar transport, chaperoning of proteins and yet unidentified roles.7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18
BBS is a heterogeneous genetic disease with variable expressivity, even within families. Families in which BBS appeared to have incomplete penetrance despite the presence of two mutations in a BBS gene were first reported in 2001 and since then multiple studies suggested that BBS with its triallelic requirement for penetrance is a model for oligogenicity that bridges the gap between Mendelian and complex disorders.19, 20, 21, 22, 23, 24, 25 Others, however, failed to demonstrate this oligogenic model and debate continues as to the magnitude of oligogenicity in the inheritance of BBS.12, 26, 27, 28, 29, 30 It is important to highlight that oligogenicity here is used in the context of penetrance (the classic all-or-none definition); otherwise, there is little doubt that epistasis is a ubiquitous phenomenon in systems biology. Indeed, it has long been realized that there is no single disease that is ‘monogenic’ in the strict sense of the word.31, 32
In order to investigate the extent to which oligogenicity contributes to the inheritance of BBS, we conducted a comprehensive genetic analysis on 29 BBS families. Our goal was not only to define the pathogenic mutations in these families, which we demonstrate can efficiently be done with the use of homozygosity mapping, but also to examine epistasis between all previously reported BBS genes and between the BBS genes and the ‘modifier’ MGC1203 (CCDC28B) that was reported to contribute to ‘oligogenic’ BBS.20 Taking advantage of the large family structure as well as the high degree of locus and allelic heterogeneity we have previously demonstrated in our consanguineous population,33 we were able to test triallelism. Our results argue against the triallelic model of BBS in the majority of cases.
Materials and methods
Human subjects
Patients were recruited through the clinical genetics program with written informed consent (KFSHRC IRB Protocol RAC#2070023). Diagnosis of BBS was made when four of the primary features or three primary features and two secondary features were present.34 If index met the diagnostic criteria, relatives with retinitis pigmentosa (RP) but who do not meet the above definition were also considered affected. All available unaffected siblings were also enrolled. All patients had thorough clinical evaluation that included medical history, physical and dysmorphology examination, anthropometric measurements, ophthalmology evaluation, random blood glucose, liver and renal function tests, abdominal ultrasound and chest X-ray. Whenever parents and/or patients were agreeable, clinical photographs were also obtained. Blood was collected in EDTA tubes from all affected and unaffected members and, with only a few exceptions, in PAXGene tubes from at least one affected member per family for DNA and RNA extraction, respectively.
Homozygosity mapping and sequence analysis
For genotyping, DNA samples were processed following the instructions provided by Affymetrix for their 250K StyI SNP Arrays (Affymetrix, Santa Clara, CA, USA). Homozygosity mapping was carried out using the CNAG tool.35 Although priority was given to BBS genes residing within runs of homozygosity (ROH), BBS1-14 genes as well as the MGC1203 (CCDC28B)-modifier gene were fully sequenced eventually in all patients and their unaffected relatives (the entire coding and flanking intronic regions up to 100 bp). We also amplified and sequenced cDNA fragments in select patients in order to confirm splice-site mutations (primers are listed in Supplementary Table S1). cDNA was also analyzed for BBS genes contained within ROH but harbored no mutation on genomic DNA analysis. Direct bidirectional sequencing was performed on 3730xL Genetic Analyzer (Applied Biosystems, Carlsbad, CA, USA). Sequences were analyzed using the Seqman II program of the DNASTAR analysis package (Lasergene, Madison, WI, USA). All sequence variants are described according to the human standard nomenclature v.2.0 (http://www.hgvs.org/mutnomen) and checked using Mutalyzer 2.0 beta-12 (https://mutalyzer.nl). The following RefSeqGene genomic and RefSeq transcript reference sequences, respectively, were used: BBS1 (NG_009093.1, NM_024649.4), BBS2 (NG_009312.1, NM_031885.3), BBS3/ARL6 (NG_008119.1, NM_177976.1), BBS4 (NG_009416.2, NM_033028.3), BBS5 (NG_011567.1, NM_152384.2), BBS6/MKKS (NG_009109.1, NM_170784.1), BBS7 (NG_009111.1, NM_176824.2), BBS8/TTC8 (NG_008126.1, NM_144596.2), BBS9 (NG_009306.1, NM_014451.3), BBS10 (NG_016357.1, NM_024685.3), BBS11/TRIM32 (NG_011619.1, NM_014010.4), BBS12 (NG_021203.1, NM_001178007.1), BBS13/MKS1 (NG_013032.1, NM_001165927.1) and BBS14/CEP290 (NG_008417.1, NM_025114.3) Intronic sequence alterations were evaluated in silico (http://www.fruitfly.org/seq_tools/splice.html) and those predicted to affect splicing were further evaluated by two-step RT-PCR. Missense mutations were verified by sequencing 96 Saudi controls (192 chromosomes), by querying the 1000 Genome database (http://browser.1000genomes.org/index.html), by checking for conservation at the protein level using the multalin software v.5.4.1 (http://www-archbac.u-psud.fr/genomics/multalin.html) as well as by checking for effect on exonic splice enhancers (http://rulai.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi?process=home) and by checking the polyphen prediction of functional effect of these variants (http://genetics.bwh.harvard.edu/pph/).
Results
BBS is clinically heterogeneous and family history can be critical for case definition
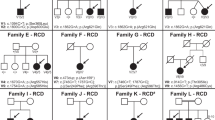
A total of 29 families were enrolled in this study, six of which were previously reported by our group.36 Table 1 summarizes the clinical features of all the families enrolled in this study (Figure 1). All but one family were consanguineous and all were of Arab origin. In general, we observed low degree of variability within families (Table 1). Interestingly, and consistent with a recent report,37 some siblings did not meet the strict definition of BBS but did have the disease mutation (see below), and hence we suggest a positive family history should serve as a primary diagnostic feature. One family deserves particular attention. In family BBS-F015, the two affected sisters of the index had RP as the only BBS manifestation whereas their index sister had all primary features of BBS so this family displays the highest degree of intrafamilial variability in our cohort. We have previously reported the first occurrence of nonsyndromic RP in patients with BBS3 mutations so this family with BBS9 mutation (see below) adds to this highly unusual BBS phenotype.36, 38 We note that RP is almost a universal feature of BBS in our cohort with the exception of BBS-F032-A probably because of her young age (2.5 years), as this BBS trait is known to display age-related penetrance.39

Clinical photographs showing variable severity of BBS manifestations. Facies range from typical round (a–c) to near normal (d). (e) Severe brachydactyly. (f) Polydactylous hand with much milder degree of brachydactyly. Note the different level of placement of postaxial polydactyly in the same individual which is also shown in (g) and (h) but in a much milder form. (i) Severe form of male hypogenitalism.
Homozygosity scan is highly effective in the molecular analysis of BBS and can guide the search for cryptic splicing mutations
We have previously demonstrated the utility of homozygosity scans in the molecular analysis of genetically heterogeneous disorders in general and BBS in particular.36, 38, 40, 41 Taking advantage of the consanguineous nature of the study cohort, we prioritized BBS genes for sequencing based on the results of homozygosity mapping. Indeed, with the exception of one non-consanguineous family (BBS-F009), all families had ROH that overlapped with at least one BBS gene, and six of these families were previously reported.36 In the remaining new 23 families, homozygosity scan allowed us to identify the disease-causing mutation by sequencing only one or two BBS genes. A total of 13 mutations were identified, all of which are novel except for the previously reported BBS10 c.728_731del, p.(K243Ifs*15) (Figure 2 and Table 2).42 In one family (BBS-F032), MKS1 and BBS10 were sequenced first as suggested by ROH analysis but no DNA alterations were identified. cDNA analysis on the other hand was unable to confirm the presence of exon 1 in the MKS1 transcript NM_001165927.1 (ENST00000537529) whereas BBS10 cDNA analysis was normal (Figure 2). Genomic sequencing of the entire intron 1 as well as the 1.5-kb region upstream of exon 1 failed to reveal any genomic variant that may explain aberrant MKS1 transcription. We note here that this exon 1 loss is not going to affect the other five known protein-coding MKS1 transcripts, which use an alternate first coding exon 1 (ENST00000393120, ENST00000313863, ENST00000393119, ENST00000337050, ENST00000546108). Thus, the exact cause of the aberrant transcription remains unclear. This observation is consistent with our previous experience with another BBS mutation which we demonstrated to be pathogenic at the RNA level and failed to identify the underlying intronic mutation but have since identified the cause as a very deep intronic mutation in BBS1 that abolishes an intronic splice enhancer sequence (see BBS-F006 in Table 2).36 What these results highlight is the importance of complementing DNA analysis with RNA analysis in patients with BBS whenever possible.

Sequence chromatogram of all 12 novel BBS mutations. For each mutation, the upper panel represents the patient sequence with the mutation indicated by red asterisk and the lower panel is for comparison with normal control. Details of the RT-PCR experiment to show the exon loss in BBS9 and BBS13 are shown. Tiling PCR fragments confirmed that BBS9 exon 6 is deleted at the genomic level as part of a 6.7–7.2-kb deletion (Supplementary Figure S1). A pie chart is shown in the bottom to summarize the relative contribution of each BBS gene to the overall mutation pool of BBS in the study population.
Locus and allelic heterogeneity in BBS is common in Arabs
The 12 novel mutations affected most of the known BBS genes (BBS2, BBS4, BBS5, BBS6/MKKS, BBS7, BBS9, BBS10, BBS12 and BBS13/MKS1). Two were nonsense mutations (BBS10 c.1365T>G, p.(Y455*) and BBS4 c.1180C>T, p.(Q394*)), one was in-frame deletion (BBS12 c.1993_1996del, p.(V665Lfs*14)) and four were frameshift mutations that predict premature truncation (BBS7 c.602-2A>T, BBS10 c.1889_1893del, p.(S630Nfs*4), BBS9 exon 6 deletion and BBS13/MKS1 exon 1 loss). Two of these frameshift mutations were caused by abnormal splicing. Thus, splicing mutations represent a major class of mutations in BBS among Arabs where six out of the 19 mutations we describe in Arabs (including our previous study36 belong to this class (31.5%). The MKS1 mutation is particularly interesting because there is only one instance in which two MKS1 alleles were demonstrated to result in BBS phenotype, and hence our finding provides additional support of MKS1 as a bona fide BBS gene.43 All missense mutations (n=5) and the one in-frame deletion replaced highly conserved amino acids except for the c.1207C>T, p.(R403C) in BBS2 in family BBS-F024. None of the mutations was identified in a panel of 96 ethnically matched controls or in the 1000 Genomes Project, and none was predicted to affect exonic splice enhancers. In order to rule out the possibility that the actual mutation is deep intronic in BBS2, we have performed RT-PCR and confirmed that the BBS2 transcribed is normally spliced. As BBS10 was the only other BBS gene (including the recently described homolog of Drosophila frtz, BBS15/C2ORF86) within ROH in this family, we sequenced this gene both at the genomic and RNA level, and identified no mutation which lends further support to the notion that p.(R403C) is possibly the pathogenic BBS2 mutation in this family. However, PolyPhen predicts this amino-acid change to be benign. Therefore, unless future functional work shows that this amino acid serves a species-specific function in the humans, we caution that this may represent a benign variant and that the actual disease-causing mutation may be in yet unidentified BBS locus.
No evidence of triallelism involving BBS genes for penetrance or expressivity
Although homozygosity mapping did assist us in quickly identifying the underlying homozygous BBS mutations in all study families (except for the one non-consanguineous family in which direct sequencing was required to identify the compound heterozygosity), our main goal in this study was to investigate the magnitude of oligogenicity in BBS. Therefore, we sequenced all remaining BBS genes as well as the ‘modifier’ MGC1203 (CCDC28B) both in affected and unaffected members of these families. In total, more than 30 000 amplicons were sequenced and analyzed. To the best of our knowledge, this is the first study that takes this comprehensive approach rather than targeted gene analysis in affected patients to address the hypothesis of oligogenicity in BBS. This massive sequencing effort, not unexpectedly, did uncover a number of novel sequence variants in BBS genes other than the BBS gene harboring the two pathogenic mutations per family (n=41, all previously unreported). The overwhelming majority of these variants were non-coding (30/41 or 73%) and with the exception of three UTR variants (BBS10 c.-52C>T, MKS1 c.-17C>G and TRIM32 c.*229C>T), the remaining were intronic that ranged in depth from the nearest exon–intron junction between 17 to >100 bp and none was predicted in silico to perturb splicing. These additional alleles, therefore, are likely to represent ethnic-specific SNPs rather than serve as third alleles. Several observations make the remaining 11 variants that did result in a change of amino acid also unlikely to act as penetrance- or even expressivity-modifying third alleles. First, and in demonstration of the advantage of studying multiplex families to address the issue of oligogenicity, we did not observe these variants consistently among affected patients with the same mutation, that is, they were not necessary to make the other two alleles penetrant. Furthermore, their presence did not correlate with increasing disease severity as estimated by the number of primary and secondary features of the disease. In particular, in family BBS-F015 which represented the extreme end of variable expressivity in our cohort and would therefore be expected to display the presence of ‘modifiers’ in the other BBS genes, we failed to show any such variant in any of the 14 genes screened. Second, these variants were found at a relatively high frequency in ethnically matched controls. Indeed, our results urge caution about unjustified labeling of variants found in other BBS genes as ‘modifiers’ because, as we demonstrate by the massive sequencing we undertook in this study, such variants are likely to be identified when many genes are sequenced merely by chance.
Several mutations are worth highlighting as examples of what may have otherwise been viewed as examples of oligogenic inheritance. BBS4 c.157-2A>G, p.(A53Hfs*2) is a mutation we previously demonstrated experimentally to completely abolish the normal transcript.36 This mutation was identified in four families. BBS-arRP-F026 is a family of four affected siblings who all harbored an additional BBS9 variant (c.1546C>A, p.(P516T)) in the heterozygous state. Index patient in family BBS-F017 also harbored an additional missense variant in CEP290 (c.2963A>C, p.(Q988P)). However, the same BBS4 mutation was also found in two families (BBS-F005 and BBS-F013), who had no evidence of any ‘third’ allele in any of the 14 genes tested and were equally affected clinically. Thus, this mutation is clearly sufficient in the homoallelic state to cause BBS and the two observed variants are likely to represent chance association. Another example is family BBS-F019 in which BBS is caused by MKKS mutation c.871G>T, p.(V291F). Our sequencing of the unaffected siblings revealed the presence of several variants not shared with the only affected member. In fact, the only missense among these variants (BBS9 c.1111G>A, p.(V371I)) was found in the two normal siblings but not in the index. Theoretically, this heterozygous variant could have been inherited by the index and raised suspicion of oligogenicity, further highlighting the need for caution to avoid misinterpreting sequence variants as modifiers.
Discussion
The argument for oligogenic inheritance of BBS is based on three proposed lines of evidence.23 First, BBS mutations are not sufficient to cause the disease in some patients, that is, normal individuals exist who harbor two pathogenic variants in a BBS gene. Second, BBS patients with two pathogenic mutations in a BBS gene harbor ‘third alleles’ in other BBS genes. Third, carrier frequency of BBS genes in the general population is higher than what would be expected for calculated disease frequency of BBS. We argue that, in many cases, these three lines of evidence have not been demonstrated conclusively in the literature. For example, with the exception of the compound heterozygosity for a truncating BBS2 mutation in the original report,25 almost all other reports of ‘non-penetrant’ BBS mutation were missense mutations. Surprisingly, despite the reported 20% frequency of the presence of ‘third’ alleles in Caucasian BBS patients, it is generally accepted that cases of non-penetrance are exceedingly rare (Katsanis, personal communication) even though non-penetrance is an essential prediction of the triallelism theory.
We acknowledge that this study involves a smaller number of families compared with some of the previous studies that suggested oligogenicity in BBS. However, we believe the unprecedented sequencing of all 14 BBS genes and the MGC1203 modifier in both affected and, as importantly, in unaffected members of mostly large families lend credence to the significance of our failure to identify a single unequivocal instance of triallelism. We stress here that, despite the private nature of most of our mutations, the remarkable degree of locus and allelic heterogeneity we observed in our population supports the generalizability of our data and argues against the hypothetical possibility that our use of a different population was the reason for the apparent lack of oligogenicity. We also acknowledge that our study design essentially misses the presence of ‘third’ alleles in yet unidentified BBS genes or other ciliopathy genes. However, the available body of literature is primarily concerned with the ‘third’ allele being one of the known BBS genes.
The findings of this study make it possible to suggest two scenarios that may have erroneously supported triallelism in BBS. The first is related to the cryptic splicing mutations, which can be very difficult to identify on DNA sequencing. Although the homoallelic nature of these mutations in this study allowed us to identify them, it is quite possible that the presence of such mutations in compound heterozygous state with other easier to identify mutations can lead to the erroneous conclusion that the BBS gene in question carries only one allele and the subsequent search for other alleles can indeed uncover some, although these may not be necessarily pathogenic as we showed. We also suggest another scenario in which extremely mild expressivity in the form of isolated RP may be interpreted as non-penetrance as this is an age-dependant phenotype.
It is noteworthy that our ability to identify the mutation in all families enrolled in this ongoing study was very helpful in directly testing the triallelism hypothesis. This detection rate is higher than what is described in other studies.29, 44 Indeed, 75% detection rate is commonly cited in the literature. Clearly, genetic homogeneity and founder effect in the study population cannot be invoked as plausible explanations to our high detection rate as they are in direct contradiction to what we observed in this study. In fact, and consistent with our theory that, in the right setting, consanguinity overrides founder effect, we have observed a similar phenomenon in BBS.33 Our experience with cryptic splicing mutations suggests that our ability to combine genomic DNA analysis with targeted transcript analysis that is informed by homozygosity scan is a likely reason for our high detection rate. In other words, although we acknowledge the potential presence of other BBS loci, we believe their presumed contribution is probably inflated by the inability to identify unusual mutations in known BBS genes in previous studies.
In conclusion, we show in the most comprehensive sequencing-based study to date on BBS that evidence is lacking of oligogenicity. Although this study cannot conclusively rule out the possibility of occurrence of oligogenic BBS, it does suggest that such occurrence, if it exists, is not seen in the overwhelming majority of BBS patients. Because of the uncertainty surrounding genetic counseling of families with this disease that stems from reports of oligogenicity, we believe this study and others that support a model in which BBS is a fully penetrant autosomal recessive disease in the overwhelming majority of cases will be of great clinical utility.
References
Harnett JD, Green JS, Cramer BC et al: The spectrum of renal disease in Laurence-Moon-Biedl syndrome. N Engl J Med 1988; 319: 615–618.
Green JS, Parfrey PS, Harnett JD et al: The cardinal manifestations of Bardet-Biedl syndrome, a form of Laurence-Moon-Biedl syndrome. N Engl J Med 1989; 321: 1002–1009.
Bardet G : On congenital obesity syndrome with polydactyly and retinitis pigmentosa (a contribution to the study of clinical forms of hypophyseal obesity). 1920. Obes Res 1995; 3: 387–399.
Biedl A : A pair of siblings with adiposo-genital dystrophy. 1922. Obes Res 1995; 3: 404.
Tobin JL, Beales PL : The nonmotile ciliopathies. Genet Med 2009; 11: 386–402.
Pedersen LB, Veland IR, Schroder JM, Christensen ST : Assembly of primary cilia. Dev Dyn 2008; 237: 1993–2006.
Ansley SJ, Badano JL, Blacque OE et al: Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome. Nature 2003; 425: 628–633.
Blacque OE, Reardon MJ, Li C et al: Loss of C. elegans BBS-7 and BBS-8 protein function results in cilia defects and compromised intraflagellar transport. Genes Dev 2004; 18: 1630–1642.
Fan Y, Esmail MA, Ansley SJ et al: Mutations in a member of the Ras superfamily of small GTP-binding proteins causes Bardet-Biedl syndrome. Nat Genet 2004; 36: 989–993.
Kulaga HM, Leitch CC, Eichers ER et al: Loss of BBS proteins causes anosmia in humans and defects in olfactory cilia structure and function in the mouse. Nat Genet 2004; 36: 994–998.
Li JB, Gerdes JM, Haycraft CJ et al: Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell 2004; 117: 541–552.
Mykytyn K, Nishimura DY, Searby CC et al: Identification of the gene (BBS1) most commonly involved in Bardet-Biedl syndrome, a complex human obesity syndrome. Nat Genet 2002; 31: 435–438.
Nishimura DY, Fath M, Mullins RF et al: Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc Natl Acad Sci USA 2004; 101: 16588–16593.
Fath MA, Mullins RF, Searby C et al: Mkks-null mice have a phenotype resembling Bardet-Biedl syndrome. Hum Mol Genet 2005; 14: 1109–1118.
Chiang AP, Beck JS, Yen HJ et al: Homozygosity mapping with SNP arrays identifies TRIM32, an E3 ubiquitin ligase, as a Bardet-Biedl syndrome gene (BBS11). Proc Natl Acad Sci USA 2006; 103: 6287–6292.
Yen HJ, Tayeh MK, Mullins RF, Stone EM, Sheffield VC, Slusarski DC : Bardet-Biedl syndrome genes are important in retrograde intracellular trafficking and Kupffer's vesicle cilia function. Hum Mol Genet 2006; 15: 667–677.
Tayeh MK, Yen HJ, Beck JS et al: Genetic interaction between Bardet-Biedl syndrome genes and implications for limb patterning. Hum Mol Genet 2008; 17: 1956–1967.
Davis RE, Swiderski RE, Rahmouni K et al: A knockin mouse model of the Bardet-Biedl syndrome 1 M390R mutation has cilia defects, ventriculomegaly, retinopathy, and obesity. Proc Natl Acad Sci USA 2007; 104: 19422–19427.
Zaghloul NA, Liu Y, Gerdes JM et al: Functional analyses of variants reveal a significant role for dominant negative and common alleles in oligogenic Bardet-Biedl syndrome. Proc Natl Acad Sci USA 2010; 107: 10602–10607.
Badano JL, Leitch CC, Ansley SJ et al: Dissection of epistasis in oligogenic Bardet-Biedl syndrome. Nature 2006; 439: 326–330.
Eichers ER, Lewis RA, Katsanis N, Lupski JR : Triallelic inheritance: a bridge between Mendelian and multifactorial traits. Ann Med 2004; 36: 262–272.
Badano JL, Kim JC, Hoskins BE et al: Heterozygous mutations in BBS1, BBS2 and BBS6 have a potential epistatic effect on Bardet-Biedl patients with two mutations at a second BBS locus. Hum Mol Genet 2003; 12: 1651–1659.
Beales PL, Badano JL, Ross AJ et al: Genetic interaction of BBS1 mutations with alleles at other BBS loci can result in non-Mendelian Bardet-Biedl syndrome. Am J Hum Genet 2003; 72: 1187–1199.
Katsanis N, Eichers ER, Ansley SJ et al: BBS4 is a minor contributor to Bardet-Biedl syndrome and may also participate in triallelic inheritance. Am J Hum Genet 2002; 71: 22–29.
Katsanis N, Ansley SJ, Badano JL et al: Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science 2001; 293: 2256–2259.
Nakane T, Biesecker LG : No evidence for triallelic inheritance of MKKS/BBS loci in Amish Mckusick-Kaufman syndrome. Am J Med Genet A 2005; 138: 32–34.
Hichri H, Stoetzel C, Laurier V et al: Testing for triallelism: analysis of six BBS genes in a Bardet-Biedl syndrome family cohort. Eur J Hum Genet 2005; 13: 607–616.
Slavotinek AM, Searby C, Al-Gazali L et al: Mutation analysis of the MKKS gene in McKusick-Kaufman syndrome and selected Bardet-Biedl syndrome patients. Hum Genet 2002; 110: 561–567.
Muller J, Stoetzel C, Vincent MC et al: Identification of 28 novel mutations in the Bardet-Biedl syndrome genes: the burden of private mutations in an extensively heterogeneous disease. Hum Genet 2010; 127: 583–593.
Smaoui N, Chaabouni M, Sergeev YV et al: Screening of the eight BBS genes in Tunisian families: no evidence of triallelism. Invest Ophthalmol Vis Sci 2006; 47: 3487–3495.
Mulvihill JJ : Craniofacial syndromes: no such thing as a single gene disease. Nat Genet 1995; 9: 101–103.
Alper JS : Genetic complexity in single gene diseases. BMJ 1996; 312: 196–197.
Aldahmesh MA, Abu-Safieh L, Khan AO et al: Allelic heterogeneity in inbred populations: the Saudi experience with Alstrom syndrome as an illustrative example. Am J Med Genet A 2009; 149A: 662–665.
Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA : New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet 1999; 36: 437–446.
Nannya Y, Sanada M, Nakazaki K et al: A robust algorithm for copy number detection using high-density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer Res 2005; 65: 6071–6079.
Abu Safieh L, Aldahmesh MA, Shamseldin H et al: Clinical and molecular characterisation of Bardet-Biedl syndrome in consanguineous populations: the power of homozygosity mapping. J Med Genet 2010; 47: 236–241.
Pawlik B, Mir A, Iqbal H et al: A Novel Familial BBS12 mutation associated with a mild phenotype: implications for clinical and molecular diagnostic strategies. Mol Syndromol 2010; 1: 27–34.
Aldahmesh MA, Safieh LA, Alkuraya H et al: Molecular characterization of retinitis pigmentosa in Saudi Arabia. Mol Vis 2009; 15: 2464–2469.
Spaggiari E, Salati R, Nicolini P, Borgatti R, Pozzoli U, Polenghi F : Evolution of ocular clinical and electrophysiological findings in pediatric Bardet-Biedl syndrome. Int Ophthalmol 1999; 23: 61–67.
Alkuraya FS : Homozygosity mapping: one more tool in the clinical geneticist's toolbox. Genet Med 2010; 12: 236–239.
Shaheen R, Al-Dirbashi OY, Al-Hassnan ZN et al: Clinical, biochemical and molecular characterization of peroxisomal diseases in Arabs. Clin Genet 2011; 79: 60–70.
Stoetzel C, Laurier V, Davis EE et al: BBS10 encodes a vertebrate-specific chaperonin-like protein and is a major BBS locus. Nat Genet 2006; 38: 521–524.
Leitch CC, Zaghloul NA, Davis EE et al: Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat Genet 2008; 40: 443–448.
Janssen S, Ramaswami G, Davis EE et al: Mutation analysis in Bardet-Biedl syndrome by DNA pooling and massively parallel resequencing in 105 individuals. Hum Genet 2011; 129: 79–90.
Acknowledgements
We thank the patients and their families for their enthusiastic participation. This study was funded by a grant from KACST to FSA (08-MED497-20). Salary support to LA was provided by a collaborative grant from DHFMR to FSA.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Rights and permissions
About this article
Cite this article
Abu-Safieh, L., Al-Anazi, S., Al-Abdi, L. et al. In search of triallelism in Bardet–Biedl syndrome. Eur J Hum Genet 20, 420–427 (2012). https://doi.org/10.1038/ejhg.2011.205
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2011.205
Keywords
This article is cited by
-
Allelic overload and its clinical modifier effect in Bardet-Biedl syndrome
npj Genomic Medicine (2022)
-
Novel biallelic variant in BBS9 causative of Bardet–Biedl syndrome: expanding the spectrum of disease-causing genetic alterations
BMC Medical Genomics (2021)
-
A novel splice site mutation in the SDCCAG8 gene in an Iranian family with Bardet–Biedl syndrome
International Ophthalmology (2021)
-
NGS: Gestern, heute und morgen
Medizinische Genetik (2019)
-
Kinesin 1 regulates cilia length through an interaction with the Bardet-Biedl syndrome related protein CCDC28B
Scientific Reports (2018)
