

Abstract
Retinal degeneration 10 (rd10) mice are a model of autosomal recessive Retinitis Pigmentosa (RP), identified by Chang et al. in 2002. These mice carry a spontaneous mutation of the rod-phosphodiesterase (PDE) gene, leading to a rod degeneration that starts around P18. Later, cones are also lost. Because of photoreceptor degeneration does not overlap with retinal development, and light responses can be recorded for about a month after birth, rd10 mice mimic typical human RP more closely than the well-known rd1 mutants. Aim of this study is to provide a comprehensive analysis of the morphology and function of the rd10 mouse retina during the period of maximum photoreceptor degeneration, thus contributing useful data for exploiting this novel model to study RP.
We analyze the morphology and survival of retinal cells in rd10 mice of various ages with quantitative immunocytochemistry and confocal microscopy; we also study retinal function with the electroretinogram (ERG), recorded between P18 and P30.
We find that photoreceptor death (peaking around P25) is accompanied and followed by dendritic retraction in bipolar and horizontal cells, which eventually undergo secondary degeneration. ERG reveals alterations in the physiology of the inner retina as early as P18 (before any obvious morphological change of inner neurons) and yet consistently with a reduced band amplification by bipolar cells.
Thus, changes in the rd10 retina are very similar to what previously found in rd1 mutants. However, an overall slower decay of retinal structure and function predict that rd10 mice might become excellent models for rescue approaches.
Keywords: retinitis pigmentosa, bipolar cell, horizontal cell, confocal microscopy, immunocytochemistry, ERG
A very high number of genetic mutations affect the eye. Those occurring in photoreceptor or pigment epithelium -specific genes often cause retinal degenerations (RDs), a family of inherited dystrophies characterized by photoreceptor dysfunction and death.
It is estimated that more than 15 million people worldwide have vision loss due to inherited forms of RD. These include patients suffering from retinitis pigmentosa (RP), a disease for which there is no cure yet (Chader, 2002).
For over 30 years, the retina of rodents has provided an invaluable tool to study the dynamics and mechanisms of inherited RD, as mouse photoreceptors undergo dystrophies caused by spontaneous DNA mutations, closely related to those of humans.
Among the mouse models, the best characterized is the retinal degeneration 1 (rd1) mouse, first described decades ago as a rodless phenotype (reviewed in Farber et al., 1994). This animal carries a spontaneous mutation of the β-subunit of the rod-phosphodiesterase (PDE) gene, causing the massive death of rods in the first weeks of postnatal life. As in typical RP, cones eventually die as well (LaVail et al., 1997; Pierce, 2001). The fast degeneration of photoreceptors is an obvious limit to the employment of rd1 mutants for rescue studies; also, the onset of rod death overlaps with the late phase of retinal synaptogenesis. This fact makes it difficult to distinguish between primary effects of rod degeneration and consequences of abnormal development of neuronal connectivity.
Recently, another spontaneous mutant has been isolated (rd10; Chang et al., 2002), which carries a re-mutation of the same rod-PDE gene, causing a later-onset degeneration of rods, followed by the death of cones. Because of a slower progressing phenotype (more reminiscent of typical human RP), the rd10 mutant has the potential to become a model of election to study the cell biology of this disease and to test therapeutic tools, as indicated by recent studies (Otani et al., 2004; Rex et al., 2004).
The purpose of the present paper is to provide a detailed analysis of the alterations occurring in the rd10 mouse retina at various times: so far, only some information is available on the time-course of photoreceptor and blood vessel degeneration in this mutant (Chang et al., 2002; Otani et al., 2004; Rex et al., 2004). We analyze the morphology and survival of inner retinal cells, and particularly of second order neurons, at various stages of the disease progression. Also, taking advantage of the relatively slow rd10 phenotype, we study the decay of retinal function by means of electroretinogram (ERG), which in the rd1 mutant can be recorded only for few days after eye opening.
In the rd10 mouse, we find profound alterations in inner retinal neurons, concomitant to photoreceptor loss, similar to those we already reported in the rd1 mouse (Strettoi et al., 2002, 2003; reviewed in Marc et al., 2003). Yet, the overall retinal structure and function are preserved for a longer period in rd10 mutants. Considering that the inner retina represents the biological platform for repair strategies designed to cure RP, such as gene therapy (Bennett, 2000; Acland et al., 2001), transplantation (Lund et al.,1997) or the application of prosthetic devices (Loewenstein et al., 2004), our findings point to the rd10 mutation as favorable substrate for testing rescue approaches.
Methods
All experimental procedures were done in compliance with the Association for Research in Vision and Ophthalmology (ARVO) Statement for the Use of Animals in Ophthalmology and Vision Research, with the Society for Neuroscience rules for the employment of vertebrate animals in research and with the laws regulating animal experimentation of the Italian Ministry of Health.
Animals
mice of the rd10 strain (on a C57Bl6/J background) and wt C57Bl6/J control mice (both from the Jackson Laboratories, USA) were used for the present study. Animals were kept in a local facility with water and food ad libitum, in a 12 hrs light/dark cycle, with illumination levels below 60 lux.
Immunocytochemistry (ICC)
Adult (1–9 months) mice were anesthetized with intraperitoneal injection of avertin (15 mg/kg) and perfused transcardially with 4% paraformaldheyde (PAF) in 0.1M phosphate buffer, pH 7.4. Young animals (10–20 days) were enucleated under anesthesia and their eyes immersion-fixed in PAF. For ICC on sections, whole eyes were sucrose-infiltrated, frozen at −20°C and serially sectioned at 14 μm on a cryostat. Sections from groups of 3 age-matched animals of the same strain were mounted on the same slide to ensure minimal differences in tissue handling and allow consistent comparisons. ICC was also performed on retinal whole mounts, upon retinal isolation from the eye cup following fixation. Primary antibodies used to label retinal cell types are synoptically illustrated in Table 1. Secondary antibodies were anti-mouse-Oregon Green 488, anti-mouse-Alexa Fluor 488 and anti-rabbit-Alexa 568. Cones were stained with Alexa Fluor 488-conjugated peanut-lectins. Nuclear staining was obtained with micromolar solutions of ethidium homodimer 2 (all fluorescent probes from Molecular Probes, Invitrogen, Milan, Italy). Tunel staining and Fluoro Jade B fluorescent histochemistry were performed according to the manufacturer’s protocols for frozen sections (Promega Italia and Chemicon International, respectively). Retinal preparations were examined with a Leica TCS-NT confocal microscope equipped with a krypton-argon laser. To compare retinal cell morphologies in wild type and rd10 preparations, the following parameters were matched: age; fixation and immunostaining protocols; retinal eccentricity (distance from the optic nerve head); magnification, pinhole size, gain and offset of the confocal microscope; thickness of extended-focus images. Files were saved in TIFF format and exported on an image workstation, equipped with Metamorph® software. Three-four littermates for each strain, for each age (P10, P20, P30, P45, 3.5, 7 and 9 months) were used for ICC on vertical sections, and reacted with the full panel of antibodies listed above. A total number of 60 rd10 and 12 wt animals were used for ICC.
Table 1.
Synoptic table of the antibodies used in the present study
| Primary antibody | Source | Host | Code | Immunogen | Recognized bands |
|---|---|---|---|---|---|
| NK3 Receptor | Abeam, Novus Biological Inc., USA | rabbit | 300-102-ab7 | 15 synthetic peptide at the C-terminus of rat NK-3 (aa 438–452) conjugated to bovine thyroglobulin | |
| Goα | Chemicon, Temecula, CA | mouse | MAB 3073 | purified bovine brain Goα | a single protein band of 39-42 kDa |
| Glutamine Synthetase (GS) | Chemicon, Temecula, CA | mouse | MAB 302 | glutamine synthetase purified from sheep brain | single band |
| Recoverin | Chemicon, Temecula, CA | rabbit | AB5585 | recombinant human recoverin | one band of 26 kDa (manufacturer’s technical information). |
| mGluR6 | Chemicon, Temecula, CA | rabbit | RA13105 | synthetic C-terminal peptide (KKTSTMAAPPQNENAEDAK) of the rat sequence | two bands of 96 and 200 kDa (possible dimer) |
| Active caspase-3 pAb | Promega, Milan, Italy | rabbit | G7481 | a peptide from the chain 29-175 of the Caspase-3 subunit (p17 fragment of human caspase-3) | |
| calbindin D clone CB-955 | SIGMA, St. Louis, MO, USA | mouse | C9848 | is derived from the CB-955 hybrydoma produced by the fusion of mouse myeloma cells and splenocytes from BALB/c mice immunized with a purified bovine kidney calbindin-D-28K. | a single 28 KDa band |
| calbindin D (EG-20) | SIGMA, St. Louis, MO, USA | rabbit | C2724 | syntetic peptide corresponding to the C terminal region of rat calbindin-D-28K (amino acids 225-244 with N-terminally added Lys-Gly) conjugated with KLH | |
| Glial-Fibrillary Acidic Protein (GFAP) | SIGMA, St. Louis, MO, USA | rabbit | G9269 | GFAP purified from human brain | |
| PKCa clone MC5 | SIGMA, St. Louis, MO, USA | mouse | P5704 | Purified bovine brain protein kinase C | 80 kDa band |
| PKCa | SIGMA, St. Louis, MO, USA | rabbit | P4334 | a syntetic peptide (Lys-Val-Asn-Pro-Gin-Phe-Val-His-Pro-lle-Leu-Gin-Ser-Ala-Val) conjugated to KLH | 80 kDa polypeptide and the minor band at 45 kDa |
| Neurofilament 200 kDa clone N52 | SIGMA, St. Louis, MO, USA | mouse | N0142 | carboxyterminal tail segment of enzimatically dephosphorilated pig neurofilament H-subunit | 200 kDa band |
Antibody specificity
NK3-R antibodies stained a population of cone bipolar cells consistent with that described in Casini et al., 2000. Goα antibody recognized in Western blot a single protein band of 39–42kDa; on retinal sections, this antibody stained ON bipolar cells, consistent with data reported by Dhingra et al., 2000. Glutamine synthetase antibodies recognized a single band of the expected molecular weight on Western blot and stained radial cells with the typical Müller glia morphology in retinal preparations. Recoverin polyclonal antibodies recognized one band of 26kDa on Western blot; they stained specifically photoreceptors and, with lower intensity, cone bipolar cells, in accordance with published results (Haverkamp et al., 2003). mGluR6 polyclonal antibody specificity was tested on Western blot; the recognised bands disappeared by antibody preabsorption with the synthetic peptide antigen. The pattern of staining was consistent with that published in Nomura et al., 1993. The antibody against Caspase-3 is specific for a cleaved form of the enzyme and recognised apoptotic cells only. In our preparations, it stained a subset of photoreceptors also labelled by TUNEL. Specificity of Calbindin D antibody was previously determined by Western blot; the staining was prevented by preabsorption with the immunizing peptide. The specificity of PKC antibodies was determined by immunoblotting. They labelled typical rod bipolar cells, as shown in Strettoi et al., 2002. The specificity of GFAP antibodies was confirmed with immunoblotting on brain extracts. Finally, the specificity of neurofilament antibody has been assessed by immunoblotting. The pattern of staining (horizontal cell axonal arbors and ganglion cells) was consistent with previous literature (Peichl et al., 1994).
Cell counting
Retinal whole mounts stained with antibodies against PKC and Calbindin D were used to estimate the number of rod bipolar and horizontal cells in rd10 mouse retinas, aged 1.5, 3.5, 7 and 9 months. Four retinal pairs were used for each age. Serial optical sections were obtained at 1 μm intervals to cover the thickness of the entire inner nuclear layer (INL), including the cell bodies of rod bipolar cells and horizontal cells. Scanning areas were 32 fields, 125 x 125 μm for rod bipolar cells and 250 x 250 μm in the case of horizontal cells. Fields were regularly spaced along the dorsoventral and nasotemporal retinal meridians. Counts of rod bipolar cells were performed on serial reconstructions covering the entire thickness of the INL, by stacking a series of single focal planes, with the aid of Metamorph® software. Each cell was identified by the site of emergence of the primary dendrite and counted in a single focal plane; each cell was then followed across the z stack and counted only once. This method does not require additional corrections to compensate for overcounting associated to cell counting procedures in single sections (Guillery, 2002).
Horizontal cells were counted in extended-focus image of the INL, corresponding to an average thickness of 12 micrometers on the z plane. Since horizontal cell bodies are largely spaced and do not overlap, counting on separate focal planes was not necessary. Retinal profiles contained labeled rod bipolar or horizontal cells were acquired with a microscope camera (Zeiss Axiocam) interfaced with dedicated software (Zeiss AxioVision) that allowed the calculation of retinal areas. Total number of cells were obtained multiplying average cellular densities by corresponding retinal areas. Statistic analysis (one way ANOVA) was performed with Excel 9.0
Average density values at each retinal locations were used to generate 8 x 8 squared matrices, that were then analyzed with Sigmaplot 8.0 software, to obtain retinal isodensity maps at various ages.
Electrophysiology
Mice were dark-adapted overnight and then anaesthetized as above. Body temperature was maintained at 37 °C with an electric blanket. The general conditions of the animals were monitored continuously by recording the electrocardiogram. Tropicamide (1% Sigma-Aldrich, Milan, Italy) drops were used to dilate the pupil. The cornea was kept moist with a thin layer of methylcellulose solution. At the end of each experiment, the animals were killed by anesthetic overdose.
Electroretinograms (ERGs) were recorded via coiled gold electrodes making contact with the cornea. A small gold plate placed in the mouth served as both reference and ground. Responses were amplified differentially, bandpass-filtered at 0.3–500 Hz and digitized at 0.25–0.5 ms interval by a PC interface (National Instruments LabVIEW 5.1, Milan, Italy). Responses (n= 5) were averaged with an inter-stimulus interval ranging from 20 s for dim lights to 5 min for the three brightest flashes.
Full-field stimulation was achieved via a Ganzfeld sphere, 30 cm in diameter, whose interior surface was coated with a highly reflective white paint. An electronic flash unit (SUNPAK B3600 DX; Tocad Company, Tokyo, Japan) generated a stimulus whose energy decayed in time with a τ = 1.7 ms. A short wavelength bandpass filter of 7.5 nm half bandwidth (Spindler and Hoyer GmbH u. Co. Göttingen, Germany) was used which gave a scotopic effective λ= 492 nm. Saturating a-wave responses were obtained by delivering flashes of white light whose scotopic and photopic efficacy were evaluated following Lyubarsky and Pugh (1996). Calibrated neutral density filters were used to attenuate the intensity of the flashes. The flash luminance at the corneal plane was measured in photometric units using a IL-700 photometer (International Light, Newburyport, MA, USA) with a scotopic filter (revised Z-Commission International de l’Eclairage, CIE) and taking the average of 20 flashes. The maximum flash intensity obtained by this procedure was 1076 cd/m2. The conversion from the flash intensity at the cornea to units of photoisomerizations rod −1 per flash (Φ) followed Sazik et al (2002). Our estimates were obtained by using the parameters for the mouse eye based on our measurements (pupil area: 3.2 mm2; retinal area: 18 mm2) and on the data of Lyubarsky et al. (2004). The estimated maximum retinal illuminance of 3443 scotopic td/s therefore corresponded to 5.5 x 105 Φ.
The leading edge of the a-wave was fitted to the model of the activation phase of the rod G-protein transduction cascade (Lamb and Pugh, 1992; Pugh and Lamb, 1993) according to equation:
| (1) |
where F(t) is the fraction of the circulating current normalized to its dark value, A is the amplification factor with the units of s−2/Φ, expressed in term of the gain parameters of the cascade stages and teff is inclusive of any delay contained in both the response and instrumentation. Further details are given in Gargini et al. (2004).
Light stimuli, sinusoidally modulated in time, were generated by a calibrated green LED, driven by a computer-assisted, special purpose, function generator (see Demontis et al. 2005). The stimulus contrast, defined as: [(Lmax − Lmin ) / (Lmax + Lmin )] x 100 was 85% and the mean luminance corresponded to 36 Φ. Sweeps acquired in steady state conditions were averaged and the power spectrum of the signal was computed by a Fast Fourier Transform (FFT) routine, implemented in Origin 6.0 (Microcal, CT).
A total number of 30 rd10 animals and 25 wt controls were used for electrophysiology experiments.
Results
1. Photoreceptor degeneration
Ethidium nuclear staining on retinal vertical sections shows the progressive decay in the number of rows in the outer nuclear layer (onl; Fig. 1). These are 12–14 at postnatal day 10 (P10) and 11–12 rows at P20. From P20 to P25, the onl drops abruptly to 2–3 layers of nuclei. At P30, Peanut-lectin (PNA) staining shows residual cones with shortened outer and inner segments and deformed shape. At P45, only a single layers of photoreceptors is left. PNA staining demonstrates that the remaining cells are mostly aberrant cones (Figure 1).
Figure 1.

Vertical sections of the rd10 mouse retina, illustrating the progressive degeneration of photoreceptors. Red signal: ethidium nuclear staining; green signal: PNA staining of cone inner segments and synaptic endings in the opl.
A. At P20, many picnotic nuclei are visible in the central retina, where photoreceptors are actively dying (arrows). At the same age, photoreceptor death at the periphery is slightly behind (arrows in B). C and D: In the following 10 days, photoreceptor number drops continuously; the onl is reduced to 3 rows in the central (C) and to 5–6 rows in the peripheral retina (D). Cones are still clearly visible, though their outer and inner segments appear short and distorted. E and F: late stages of photoreceptor degeneration; PNA staining at this late stage is inadequate to reveal cones. At P45, only one row of nuclei persists in the onl (E). At 9 months, the row is discontinuous (F). Bar is 20 micrometers.
Scattered profiles in the outer plexiform layer (opl) can be revealed in animals as old as 5 months by fluorescent PNA staining. These profiles correspond to the leftover of degenerating cones, whose pedicles persist after the loss of the outer and inner segments. Nuclear staining shows ovoidal profiles in the onl, as late as 9 months of age (Figure 1). Most likely, a small population of cones survives for a long time in selected retinal areas, as already shown for the rd1 mutant (LaVail et al., 1997; Ogilvie et al., 1997).
Degeneration of photoreceptors in rd10 retinas follows a clear center-to-periphery gradient. The decrease in the number of onl rows becomes apparent in the central retina first, while degeneration in the periphery is slightly (about 2–3 days) delayed (Fig. 1).
Ethidium staining shows nuclear condensation (pycnosis) in numerous cells in the onl (Fig. 1); in addition, degenerating photoreceptors can be labeled with common markers for cellular apoptosis. Fluoro Jade histochemistry and TUNEL staining, indicative of neuronal damage and DNA fragmentation respectively, highlight condensed cellular profiles in the outer retina, with maximum staining in the P20–P25 temporal window (Figure 2). As expected, TUNEL and Fluoro Jade staining also reveal a center-to-periphery gradient: at P26, labeled profiles are 2–3 times more frequent in the central as compared to the peripheral retina. Conversely, after P30, TUNEL positive bodies are more commonly encountered in the retinal periphery.
Figure 2.
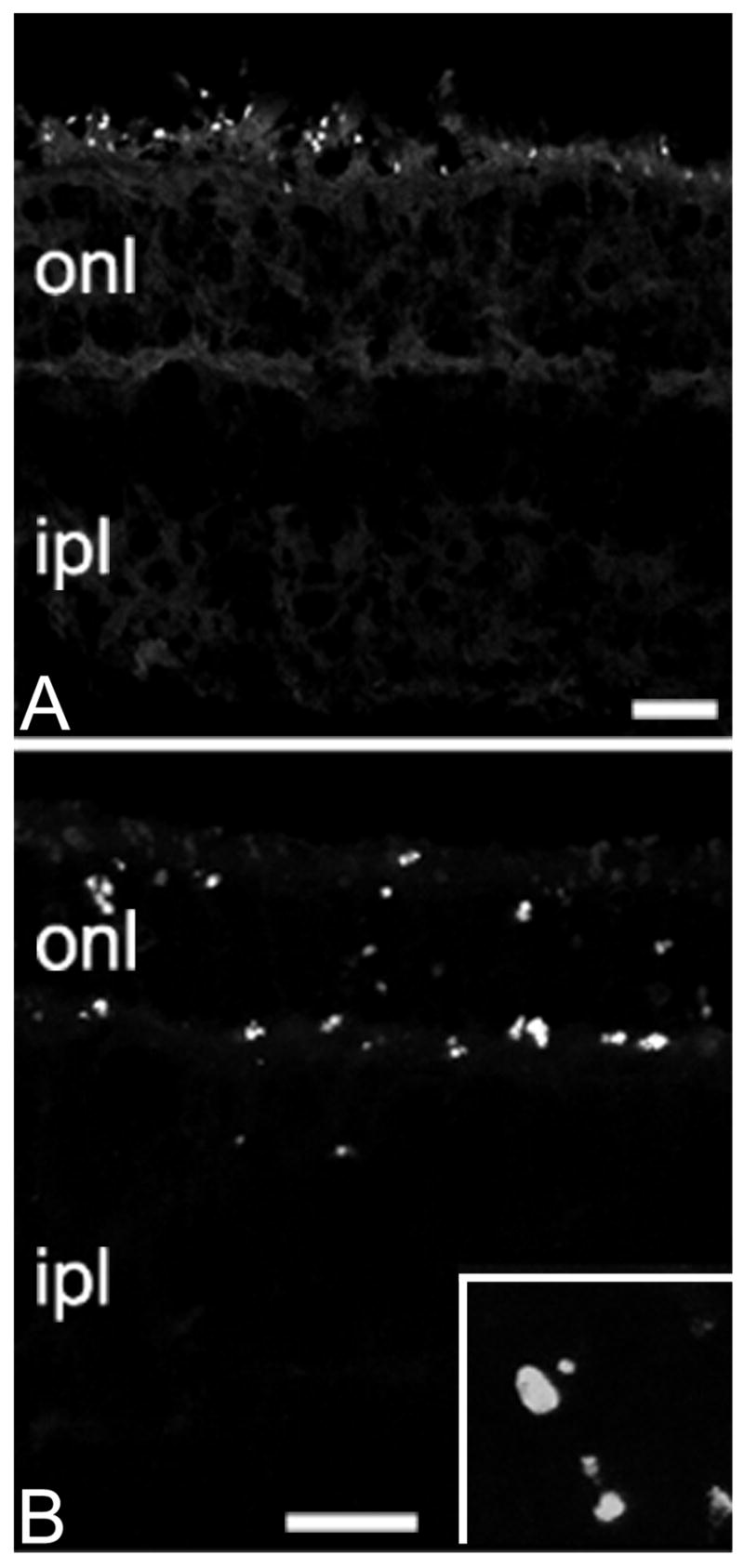
Apoptotic markers in the photoreceptor layer. A: Fluorojade-positive profiles are scattered in the outermost portion of the retina at P20. B: photoreceptors stain positively with the TUNEL method at P25. Apoptotic profiles are clearly visible (inset: 4x enlargement of the onl, similar to that shown in B). Bars are 20 micrometers.
Antibodies against the activated form of the pro-apoptotic enzyme Caspase 3 also show a specific pattern of staining in the onl, that becomes especially evident around P30. Few stained profiles, intensely labeled, are irregularly distributed throughout the onl (data not shown).
For each retinal eccentricity, the number of labeled profiles is maximum with the Fluoro Jade histochemistry, intermediate with TUNEL staining and minimum with active-Caspase 3 antibodies. This is consistent with the different markers revealed by the three methods (Schmued et al., 1997; Zeiss et al., 2004). Hence, in the rd10 mouse retina, rods start to die between the 2nd and the 3nd postnatal week; the death wave is maximum during the 4th postnatal week, about 2 weeks later as compared to the closely related rd1 mutant. Most cones disappear by 2 months of life, although a discontinuous row of nuclei persist in the onl as late as 9 months of age. Photoreceptor death seems to occur through an apoptotic mechanism, as their nuclei are picnotic, their DNA is fragmented and an activated form of Caspase 3 is expressed in some of them.
2. Rod bipolar cells
No evident alterations in the morphology and lamination of rod bipolar cells are evident in the time interval P10–P20. These neurons exhibit the well-known morphology, characterized by an elliptical cell body, about 10 μm in diameter, a bushy, chandelier-like dendritic arborization and a long axon, terminating with large varicosities in the deeper stratum of the inner plexiform layer (ipl). Rod bipolar cells are uniformly stained with anti-PKC α antibodies. Around P25, the dendrites of rod bipolar cells become shorter; this process is more apparent in the central retina (Fig. 3). In parallel to the degeneration of photoreceptors, dendritic regression progresses further and becomes obvious at P30. At P45, few dendrites are left over the rod bipolar cells of the peripheral retina, while the central-most neurons are totally devoid of processes (Fig. 4). There are no evident changes in the morphology, size and complexity of the axonal arbors of rod bipolar cells in the ipl (Fig. 3 and 4).
Figure 3.
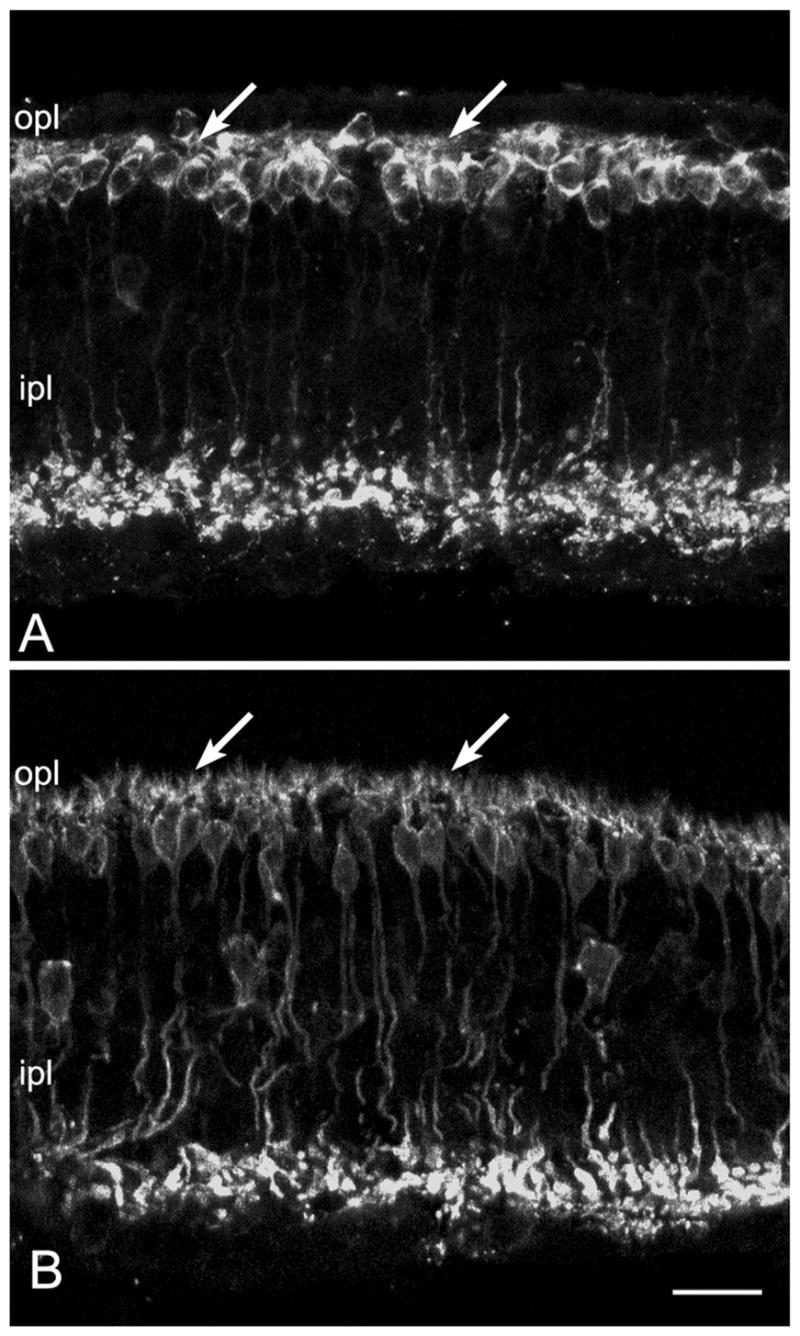
In the retina of rd10 mice, rod bipolar cells show a clear reduction in their complement of dendrites at P30 (arrows in A), compared to their wild-type counterparts (arrows in B). Bar is 20 micrometers.
Figure 4.
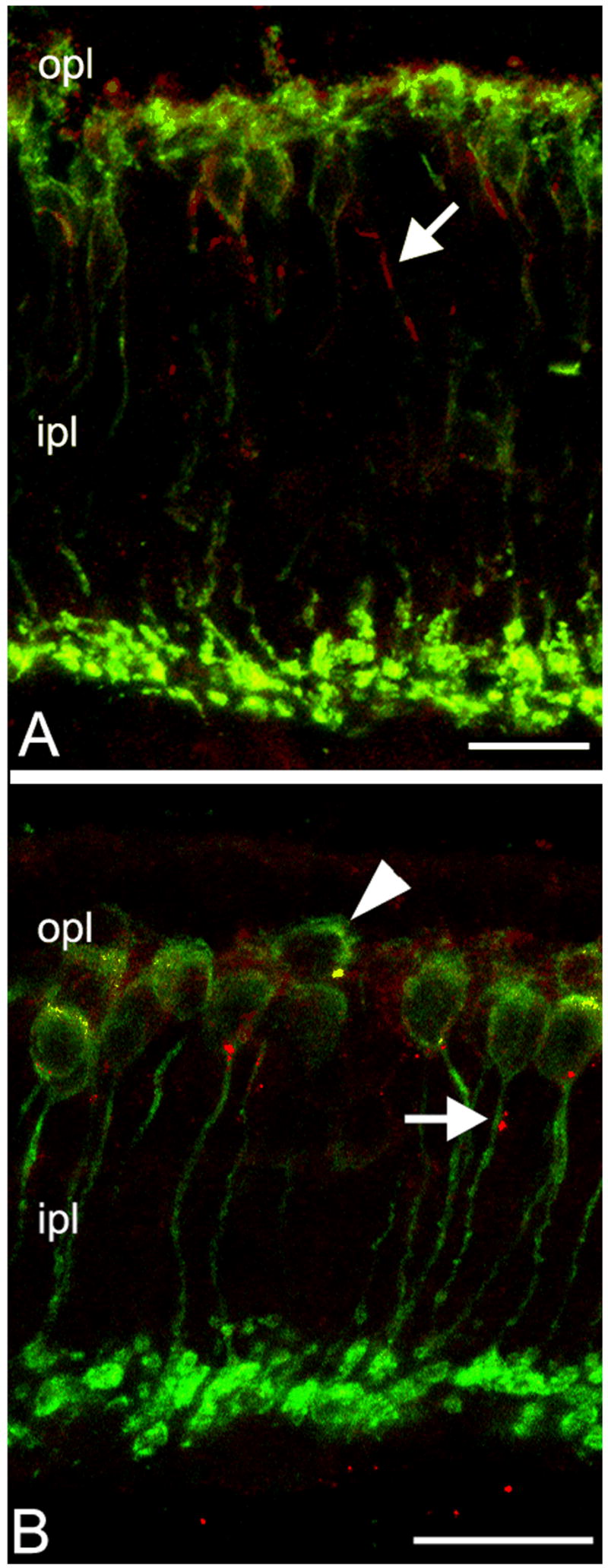
PKC staining (green) of rod bipolar cells shows the progressive retraction of dendrites, that are completely lost between P30 (A) and P45 (B, arrowhead). mGluR6 aggregates (red labeling) also decrease with time and become displaced to the cell body membrane and axons of bipolar cells (arrow). Residual clusters in the opl are mostly localized onto the dendritic tips of cone bipolar cells.
Bars are 20 micrometers.
Regression of dendrites is accompanied by changes in the distribution of mGluR6, the major glutamate receptor responsible for rods-to-rod bipolar cells synaptic communications (Fig. 4). Normally, this receptor is selectively and abundantly expressed in the opl, where it decorates the apical portion of dendrites of those bipolar cells which respond to light with a graded depolarization (rod and “ON” cone bipolar cells). Staining of lightly fixed retinal sections with mGluR6 antibodies reveals typical clusters of puncta, corresponding to receptor aggregates on the bipolar postsynaptic membrane in the opl.
The staining pattern for mGluR6 is relatively normal in retinal sections of rd10 mutant mice, up to P25, although the staining is not restricted to the tips of growing bipolar dendrites as in the wt counterpart, but also present in the cell body membrane. From P25 on, the staining in the opl decreases progressively, while an increasing number of puncta are detected in the cell body membrane, as well as in the cell axons, along their course in the inl (Figure 4A). Decrease in immunoreactivity and displacement of mGluR6 progress with time; at P45, residual staining in the opl is not associated with the dendrites of rod bipolar cells (Fig. 4B); most likely, mGluR6 clusters are linked to the dendrites of ON cone bipolar cells. Hence, together with the dendrites of rod bipolar cells, their major glutamatergic receptor has undergone a concomitant decrease and misplacement.
3. Cone bipolar cells
The mouse retina has at least 9 types of cone bipolar cells (Ghosh et al., 2004; Pignatelli and Strettoi, 2004); as these neurons are similar and have closely related molecular determinants, antibodies allowing labeling of each type individually are not available. The antibody against the receptor for the peptide Neurokin 3 labels a large number of cone bipolar cells with axonal arbors in the outermost sublamina of the ipl and flat dendrites in the opl (Casini et al., 2000). These are putative OFF-cone bipolar cells. We performed NK3-R antibody staining of vertical sections of rd10 mouse retinas from P10 on, to establish whether the staining pattern was normal. We found no apparent abnormalities in the pattern of NK3-R labeling up to the age of P45. Labeled cells formed a regular layer with bushy axonal arbors throughout the outermost half on the ipl. The dendrites were flat and clearly defined in the opl (Fig. 5). At around two months of life, the dendritic plexus started to become gradually discontinuous in the opl; animals of 4–5 months of life showed a sensible retraction of dendritic branches in NK3-R positive cells. However, we found that in animals older than 2 months, the NK3-R antibodies did not work in a reproducible manner, and then we used recoverin antibodies that label cone bipolar cells of similar morphology (Ghosh et al., 2004; Pignatelli and Strettoi, 2004). Recoverin was particularly useful after extensive photoreceptor degeneration, as only cone bipolar cells were stained and their dendrites could be visualized clearly. At 9 months of age and older, recoverin antibodies show that dendrites are virtually absent from the immunoreactive cone bipolar cells (Figure 5B).
Figure 5.
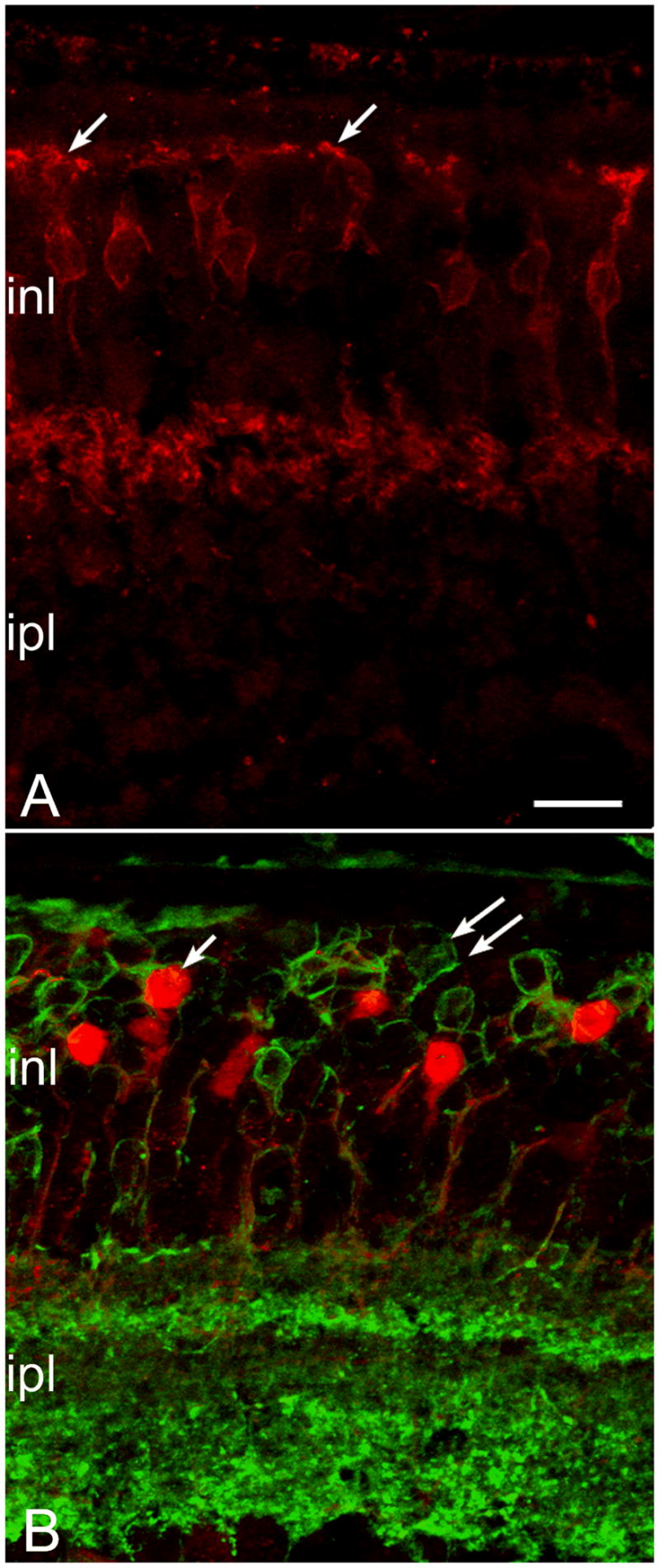
Dendritic loss from cone bipolar cells at late stages of the rd10 degeneration. A: NK3-R staining of cone bipolar cells with axonal arbors in the outer half of the ipl shows normal dendritic morphology. B: In animals 9 months and older, cone bipolar cells of the same type, labeled with recoverin antibodies (red) have totally lost the dendrites; cell bodies appear completely smooth (left arrow). Green signal: Goalpha labeling of bipolar neurons show virtual absence of processes in the opl (double arrows). Bar is 20 micrometers.
Goα antibodies label both rod bipolar cells and ON-cone bipolar cells (Dhingra et al., 2000). At 7–9 months, staining with this marker highlighted the process of dendritic retraction even more evidently. Double labeling with Goα and recoverin revealed a net decrement of the bipolar plexus in the opl (Figure 5B).
4. Horizontal cells
The rodent retina contains a single type of horizontal cell, in which the dendritic arborization arising from the cell body is postsynaptic to cone terminals in the opl. The extensive axonal arborization, connected to the cell body by means of a thin, horizontal axon, is postsynaptic to rods. The whole horizontal cell plexus can be revealed by Calbindin D immunostaining; neurofilament antibodies, instead, label the axonal arborizations (Peichl, Gonzales-Soriano, 1994). We used both antibodies to visualize horizontal cells of the rd10 retina of various ages.
The morphology of horizontal cells did not exhibit major alterations in the time interval from P10 to P45. Around two months of age, the axonal arborizations of horizontal cells, in central retinal areas, in the proximity of the optic nerve head, changed subtly, loosing the fine, end terminal processes emerging from higher order branches. With time, the gradual attenuation of the fine plexus was more evident; at 3 month of age the plexus stained by neurofilament antibodies in the opl appeared thinner and less complex. Simultaneously, the thicker processes belonging to the axonal arborizations became more visible; they formed a looser meshwork in the opl compared to the wt counterparts (Figure 6).
Figure 6.

Calbindin (red signal) and neurofilament (green) staining highlight the loss of complexity of the dendrites and axonal arborizations of horizontal cells in the rd10 mouse, at P30 (A), compared to the wt counterpart (B). The fine dendritic branches of the wt horizontal cells (arrows), are completely absent in the mutant. Bar is 20 micrometers.
Retinal whole mounts stained with the same antibodies revealed the wide-spread nature of the dendritic retraction process in horizontal cells (Figure 7). Both the dendritic and the axonal complements of fine branches went completely lost by 4 months of age. We did not detect a clear temporal mismatch in the loss of dendritic versus axonal processes from horizontal cells in the time period under study. In addition, the process of neurite sprouting from the axonal arborizations of horizontal cells, previously described in the retina of other mutants, was virtually undetectable in rd10 mice.
Figure 7.

Calbindin staining of retinal whole mounts shows the massive loss of horizontal cell processes in the retina of rd10 mice (A), compared to a wt type (B). Age (7 months), eccentricity (dorsal periphery) and thickness of the projection images are matched. Bar is 20 micrometers.
5. Müller cells
Müller glial cells were studied in the retina of rd10 mice by means of antibodies directed against the enzyme glutamine synthethase (GS), catalyzing the transformation of glutamate into glutamine, and antibodies against cytoskeletal fibrillary components, GFAP, which are known to become upregulated in glial activation.
GS antibodies suitably show the typical morphology of Müller cells starting from P20. At younger ages, these antibodies do not function properly, possibly as a low level of expression of the GS enzyme. After P20, throughout 9 months of age, GS staining of the rd10 retina was always normal and regularly distributed over the retinal surface. Only the decreased length of the outer portion of the Müller cells was evident, clearly associated to the thinning of the outer retina, caused by the massive degeneration of the photoreceptors.
In a normal, healthy retina, GFAP is expressed in astrocytes and in few Müller cells, at the far periphery of the eye. In the rd10 retina, this wt-like expression was restricted to the period P10–P20. Concomitantly with the peak degeneration of photoreceptors, right after P20, an increasing number of Müller cells appeared GFAP-positive (Figure 8). The antibody staining revealed a pattern similar to that of GS, but with a more intense labeling of the Müller cell endfeet. We detected GFAP positive processes, clearly belonging to Müller cells, in the central retina of rd10 animals, 14 months old. It is likely that GFAP in Müller cells remains high throughout the animal’s life, possibly also in relation to major vascular alterations occurring in the inner retina of the mutant (Otani et al., 2004).
Figure 8.

Up-regulation of GFAP in Müller cells of rd10 retinas aged P20 (A) and P60 (B). Note the reduction in the retinal thickness from A to B. Bar is 20 micrometers.
6. Survival of rod bipolar and horizontal cells
Rod bipolar cells
We estimated the survival of rod bipolar neurons, in the rd10 mutant, by counting PKC-positive cells in whole mount preparations from mice aged 1.5, 3.5, 7 and 9 months (Figure 9, top panel). A decrease in the number of these neurons was previously described in the central retina of rd1 mice, aged 3 months (Strettoi and Pignatelli, 2000).
Figure 9.

Histograms illustrating the survival of rod bipolar and horizontal cells in rd10 retinas aged 1.5, 3.5, 7 and 9 months, respectively. Columns are average total number of cells/retina, with standard errors. Asterisks indicate significative differences between the number of cells of each age group and that found at 1.5 months. Data were obtained by 4 independent experiments performed in triplicate and analyzed with one way ANOVA (** is for p<0.05 and *** for p<0.01).
At 1.5 months of age, rod bipolar cells were normally distributed over the retinal surface, with a typical center-to-periphery gradient, readily visible on the isodensity map (Figure10, left panel). Between 1.5 and 3.5 months of age, we found a 20% decrease in the number of rod bipolar cells. At subsequent ages, the total number of cells remained relatively stable, reaching a minimum at 9 months of age, corresponding to a 23% loss (Figure 9, top panel).
Figure 10.

Isodensity maps of rod bipolar (left) and horizontal cells (right) of the rd10 retina, at 1.5 (top) and 3.5 (bottom) months of age. Each map is based on the average densities of 4 different, age-matched retinas. Legends are cells/mm2. Note the net decrement in the density of rod bipolar cells in the nasal retina; horizontal cells, instead, decrease more rapidly in the temporal retina. At 3.5 months, the survival maps for the two cell types are almost symmetric along the dorsoventral axis.
Isodensity curves showed the anisotropy in the loss of rod bipolar cells: superimposed to a centre-to-periphery gradient, we observed a more pronounced decrease in the number of cells in the nasal quadrant of the retina (Figure 10, left panel).
Horizontal cells
We estimated the number of horizontal cells in rd10 mice of 1.5, 3.5, 7 and 9 months of age on retinal whole mounts treated with Calbindin D antibodies. As for rod bipolar cells, we observed a net loss (around 19%) of horizontal cells in the time interval comprised between 1.5 and 3.5 months (Figure 9, bottom panel). The number of cells continued to decrease with time; at 9 months, cellular loss was around 29%. Isodensity curves showed the anisotropy in the loss of horizontal cells: superimposed to a centre-to-periphery gradient, the decrement appeared more pronounced in the temporal quadrant of the retina (Figure 10, right panel).
7. Other effects
The examination of retinal whole mounts for cell counting revealed other elements of structural remodeling associated with late stages of photoreceptor degeneration. We found a clear misplacement of the cell bodies of several rod bipolar cells toward the outer retina; these cells appeared totally devoid of dendrites and formed a second layer, external to that of cells normally placed in the inl. At times, the outer row of cell bodies reached the outer limiting membrane. Sometimes these cells could also be observed in vertical sections (see Figure 4). The progressive loss of dendrites and the clear thinning of the dendritic focal plane in the opl could be optimally estimated in retinal whole mounts stained with PKC or calbindin.
At 9 months of age, staining with PKC antibodies showed a patterned disorganization of the inner retina, with bipolar cells arranged in circular domains strongly reminiscent of retinal rosettes (not shown). These patterns are characteristics of other forms of retinal degeneration (Marc et al., 2003). Rosettes, occurring in clusters over the retinal surface, were mostly revealed by the staining of rod bipolar axons and axonal arbors in the ipl and were not very evident in whole mount retinas stained with calbindin antibodies. Cell bodies of horizontal cells, however, appeared distributed over the retinal surface in a very irregular mosaic, with clear empty zones. Thus, although the observed cellular loss was overall limited, reorganization of cell bodies of inner retinal neurons did occur at late stages in the retina of rd10 mice.
8. Electrophysiology
The functional properties of the rd10 retina were explored by analyzing full field electroretinogram responses (ERG) to flashes of light, as well as to periodic stimuli, consisting of sinusoidal modulation in time of a mean luminance. Animals were ERG traced over different ages and the response from rd10 was compared with that recorded from age-matched wild type animals.
ERG responses could be recorded from rd10 mice as early as P14 up to P28. After that age, responses became sluggish and eventually undetectable. Because retinal degeneration in rd10 starts in the central retina and progresses towards the periphery, and considering that the main contribution to the full field ERG originates in the periphery, it follows that residual ERG responses may be detected up to very late stages of the degeneration.
Averaged responses to dim, intermediate and bright light flashes from wild type and rd10 mice, aged P18 and P26, are illustrated in Fig. 11. Both a-and b- waves of the mutant are delayed, slower and reduced in size compared with those of the wild type. Furthermore, the response amplitude decreases with the rd10 mouse age. All these features may be attributed to the progression of a degeneration which occurs maximally between P20 and P25 and mainly affects rods, whereby the number of visual cells contributing to the mass ERG response is substantially reduced; presumably, also contributions from individual visual cells have altered kinetics. Hence, it is expected that both a- and b- wave become slower and reduced in size, thus reducing the overall sensitivity to light of the rd10 retina.
Figure 11.

Flash responses to three different luminances in scotopic conditions, from rd10 mice (P18: blue line; P26: red line) and from P25 wild type mice (black line). (Φ= photoisomerizations rod −1 per flash; dim: Φ=40; intermediate: Φ = 15000; bright: Φ=38000).
Amplitude and time to the peak of the scotopic ERG b-wave are plotted in Fig. 12 as a function of the flash intensity for P26 wild type mice and for P18 and P26 rd10 mutants. The response amplitude of the mutants is smaller than that of wild type mice for all the intensities used. By contrast, differences in the time to peak between control and rd10 mice are prominent in responses to dim flashes but disappear in those to bright stimuli. A possible explanation for this finding is that, in the present experimental conditions, bright flashes are likely to elicit responses also from cones, whose contribution may become substantial especially when most of the rods have degenerated. This possibility is supported by the morphological finding that, between the age of P20 and P28, when retinal degeneration has already affected a large part of rods, cones are still largely spared in the rd10 retina.
Figure 12.


Amplitude and time-to-peak of scotopic ERG b-wave components, plotted as a function of stimulus intensity. Illustrated are: P25 wt mice (black line; n=8); P18 rd10 mice (blue line; n=5); P26 rd10 mice (red line; n=5). Data were analyzed with one way ANOVA (* P <0.05; ** P<0.001).
The a-wave of the ERG recorded in response to a brief flash has been widely used for a quantitative assessment of rod photoreceptor function in health and disease (Breton et al. 1994). In the present study, the leading edge of the a-wave was fitted to the model of the activation phase of the rod G-protein transduction cascade (Lamb and Pugh 1992) according to equation : F(t)=exp[−1/2 ΦA(t−teff)2] where F(t) is the fraction of the circulating current normalized to its dark value, A is the amplification factor with the units of s−2/Φ expressed in term of the gain parameters of the cascade stages and teff is inclusive of any delay contained in both the response and instrumentation. The parameter A, defined as amplification factor, is especially relevant to the analysis of rd10 responses from rods because it lumps together three distinct parameters involved in the activation steps of phototransduction, two of which are related with the PDE activation. The results of this analysis are summarised in Fig. 13. In A and B, a-wave responses to flashes of increasing intensity of wild type animals are compared with responses from rd10 mice. The dotted line superposed on the response traces has been obtained by fitting the leading edge of the a-wave to yield two parameters of the a-wave, the amplification parameter (A) and the effective delay (teff), as shown in Figure 14. It can be observed that the amplification factor is severely reduced in responses from rd10 mice and that the total delay is substantially increased.
Figure 13.

Recordings from wt mice (A) and from P21 rd10 mice (B), normalized to the saturating a-wave response. The dotted red line, superposed on the response traces, has been obtained by fitting the leading edge of the a-wave according to the equations reported in Methods. Stimulus intensities are Φ= 2.400; 15.000; 38.000; 143.000 for wt animals; and Φ= 2.400; 15.000; 38.000 for rd10 mice.
Figure 14.

Average values for two parameters of the a-wave: the amplification parameter (A) and the effective delay (teff), measured at 15.000 photoisomerization rod −1. For both wt and rd10 mice, measurements were performed on 5 animals. (** P<0.001). Bars are S.E.M.
The kinetics of the ERG response were further evaluated by using light stimuli consisting of sinusoidal modulation of a mean luminance over a range of temporal frequencies. This approach is particularly suitable for analyzing the kinetics of the b-wave which reflects mostly the post-synaptic activity of the ON-bipolar cells. Responses to sinusoidal stimuli are in fact a composite response whose single components add independently at the electrode, but the contribution from ON-bipolar is prominent. In Fig. 15 the response amplitude to sinusoidal modulation of the mean luminance is plotted as a function of the temporal frequency of the stimuli (attenuation characteristics). It appears that, while the responses to low frequency stimuli (up to 1Hz) are similar in both wild type and rd10 mice, responses to stimuli frequencies in the range 1–10 Hz of the rd10 are substantially smaller than in controls. Inspection of the attenuation characteristics also indicate that the overall trend of those from rd10 is reminiscent of a low-pass filtering, while in the wild type they are more similar to a band-pass filter.
Figure 15.

Sinusoidal ERG. Blue and red lines refer to rd10 mice, aged P18 and P26, respectively. Black lines refer to wt, age-matched animals. Gray lines represent the sinusoidal light stimulus.
Left panel: sinusoidal ERG responses to 4 different temporal frequencies.
Right panel: Kinetic analysis of ERG responses, obtained from rd10 mice aged P18 (A; n=8) and P26 (B; n=8) and from age-matched wt mice (n=8, black lines), using light stimuli sinusoidally modulated in time, from 0.3 to 30 Hz. The amplitude of the response, plotted as a function of the temporal frequency of the stimulus (attenuation characteristics), appears suppressed in the frequency range of 2–10 Hz.
Discussion
This paper provides a follow-up study of the general retinal organization in a recently isolated strain of spontaneous mutant mice with the potential of becoming an excellent model to study the biology of human retinitis pigmentosa. In rd10 mice, rods degenerate maximally between P20 and P25, in a clear centre-to-periphery gradient. Secondary degeneration of cones proceeds in parallel at a slower rate; a scattered population of aberrant cones persists up to at least 9 months of age. These findings agree with, and significantly extend, previous studies based on this mutant (Chang et al., 2002; Otani et al., 2004).
As in other models of retinal degeneration (Chang et al.1993; Remè et al., 1998), the death of photoreceptors in the rd10 mouse shows the hallmarks of apoptosis: nuclei in the onl are strongly picnotic and are stained with Tunel and Fluoro Jade methods; some of them express a form of activated Caspase 3. Apoptotic markers appear in the retina in a centre to periphery gradient.
This paper is mainly focused upon secondary changes affecting inner retinal cells. Modifications in their morphology become detectable at around one month of age, and are more widespread at 2 months. Mostly, changes affect rod bipolar and horizontal cells. Their dendritic complement becomes progressively more scant and disorganized, eventually to regress completely. Cone bipolar cells follow, at a slower rate. Dendritic retraction in bipolar cells which carry mGluR6 receptors is preceded by a decrease in the immunoreactivity for this marker, and by its displacement to the cell bodies and bipolar cell axons in the inl. In the rd10, photoreceptors start to die past the late phases of retinal synaptogenesis (Sharma et al., 2003), and die out more slowly in comparison to the rd1 counterpart: this results into a proper development of the dendrites of second order neurons. Only after extensive photoreceptor death do the dendrites of rd10 bipolar cells exhibit a retraction, possibly because of lack of trophic messages and local cues (Leveillard et al., 2003).
At present, retraction of dendrites from the opl has proven to occur in rd10 and rd1 mutants, in crx-null mice and in other rodent models of retinal degeneration (Jones et al., 2003; Cuenca et al., 2004; Wang et al., 2005). Preliminary results by our group suggest that this occurs in human RP as well (Strettoi et al., 2005). Thus, this form of remodeling can be considered as a hallmark of the secondary effects of photoreceptor death, independent of the underlying genetic defect and the affected species.
Similarly to what observed in the rd1 mutant, the morphological changes in bipolar and horizontal cells occurring in the rd10 retina are followed by their death: survival of rod bipolar and horizontal cells drops right after the peak of photoreceptor degeneration, and decreases slowly over the following months. Subsequently, the inl disorganizes, and typical rosettes appear. Other cell types, studied but not shown here, such as AII and cholinergic amacrine cells, do not seem to be affected in the first 5 months of life. A morpho-functional study of the cone pathway and of ganglion cell alterations in this mutant is in progress in our laboratories.
All along the period of active photoreceptor degeneration, and particularly between P20 and P30, as well as at later stages, GFAP is upregulated in Müller cells. As in most retinal pathologies, a glial reaction with intermediate-filaments hypertrophy accompanies and follows the death of photoreceptors (reviewed in Marc et al., 2003).
All together, the rd10 mutation appears milder as compared to the aggressive rd1, as the rd10 inner retina is more resistant to remodeling; yet, ultimately, secondary modifications are remarkably similar in the two mutants and recall patterns of alterations described in other models of photoreceptor disease (Milam et al., 1998; Jones et al., 2003; Cuenca et al., 2004; Wang et al., 2005). Evidently, a stereotyped series of events is associated to the loss of photoreceptors. A brief comparisons of the major events occurring in the retina of rd1 and rd10 mice at various times is reported in Table 1.
Several morphological changes described here have a clear physiological correlate. The observed differences between the flash ERG of normal and rd10 mice are consistent with a severe damage mainly affecting the distal rod pathway. Assuming then that the electrical field generating the a-wave is directly proportional to the suppression of the dark current by light (Breton et al., 1994), the present data indicate that the dark current is reduced in rd10 rods. The loss of functioning rods which occurs in the rd10 mutants between P20 and P30, largely contributes to the drastic reduction of the normalized maximal a-wave. This, however, may also indicate a concomitant decrease in the responsiveness of the residual rods, as confirmed by the analysis of the amplification factor, that is substantially lower in rd10 mice. These observations are consistent with the idea that in rd10 mutants, one or more of the activation steps, contributing to the amplification parameter (A), have a reduced gain. Given the nature of the rd10 mutation, it seems plausible to suppose that the catalytic activity of the rod-specific PDE is reduced. This notion is reinforced by the reduced value of the saturation rate.
The ERG measurements obtained by using a sinusoidal modulation of the mean luminance confirm, and extend, the results of the flash ERG. The observation that the attenuation characteristics of the rd10 match those of the normal mouse at low temporal frequencies but are much smaller at intermediate and high frequencies, may provide an indication that, even at the earliest stages of the degenerative process, bipolar neurons show signs of damage. This idea is based on previous studies on cats, in which the various ERG components were isolated pharmacologically. It has been shown that the attenuation characteristics of the PIII (photoreceptor component) conform to a low pass filtering, while the PII, which is thought to reflect the activation of bipolar neurons, rather behaves as a band-pass filter (Gargini et al. 1999). The present results are consistent with the idea that rod degeneration is associated with functional changes in bipolar cells, as also shown with single-cell electrophysiological experiments (Varela et al., 2003).
It is interesting to note that alterations in the shape and relative amplitude of the b wave are detected as early as P18; at this age, no major morphological modifications occur in the inner retina. Only the active degeneration of photoreceptors is evident. Thus, ERG changes in the b wave precede the detection of main morphological abnormalities in the inner retina and occur well before the extensive loss of dendrites from the opl. A change in the efficiency of the synaptic communication of rod to rod bipolar cells could explain the alterations found in the ERG using sinusoidal stimuli. Most likely, the net decrement of mGluR6, detected by immunocytochemistry at about one month of age, begins gradually days before, albeit it cannot be revealed by immunocytochemical methods. Such a decrement could result in a lower sensitivity of bipolar cells to glutamate released from photoreceptors, ultimately producing the changes in the b wave of the ERG reported here. Obviously, it is possible that other molecules not examined here, including ionic channels or second messengers in the bipolar cell dendritic machinery, crucial to the communication in the outer retina (Vardi et al., 2002), had been lost as well. The information that the changes in the b wave reported here are among the earliest signs of a perspective retinal decay, appears in itself relevant: these changes might be important for a timely diagnosis of the disease, thus confirming the importance of ERG assessment for RP identification (Fleischhauer et al., 2005).
The fact that a different mutation in the same PDE gene mutated in the rd1 mouse results in a slower and milder phenotype, demonstrates critical factors overall determining the outcome of the pathology: the slower phenotype is likely to be caused by a delayed death of photoreceptors. As for other mutations of the pde6b gene (Hart et al., 2005), the missense rd10 mutation ensures retention of a residual PDE activity. Thus, it is conceivable that cGMP accumulation and calcium raise, both associated with a defective PDE (Bowes et al., 1989, 1990) reach toxic levels in a longer time interval. Also, the metabolic overload taking place in mutated photoreceptors to extrude the excess calcium with active pumping is likely to be less intense in rd10 retinas. Hence, photoreceptor death starts later and persists for a longer period compared to the rd1.
Overall, the rd10 mutant is a good model of typical RP in humans and represents an excellent background for rescue studies. Indeed, rd10 mice have been used for stem cell transplantation and pharmacological treatments (Otani et al., 2004; Rex et al., 2004).
An important indication emerging from the present study is that, given the relatively slow occurrence of secondary events in the inner retina of the rd10 mutant, it would be significant to use this model to test restoration strategies after the end of rod degeneration; this would mimic more closely the human conditions, as possible intervention on human patients is usually conceived when large fractions of photoreceptors have already degenerated.
Table 2.
Qualitative comparisons of main retinal changes observed in rd1 and rd10 mutant mice, as a function of time. Data are from the present paper and from References quoted in the text.
| Mouse Strain | Mutation and inheritance | Degeneration features | Onset of rod degeneration | Recordable ERG Responses | Alterations in mGluR6 Distribution | Dendritic retraction in Rod Bipolar cells | Occurrence of Rod Bipolar & Horizontal cell death | Dendritic retraction in Cone Bipolar cells |
|---|---|---|---|---|---|---|---|---|
| rd1 | rod-specific PDE (β subunit) NONSENSE
Autosomal recessive |
rod-cone
center-to-periphery gradient |
P8 | P14-P16 | P18- | P10-
(possible arrest in growth) |
YES
(data available at P90 only) |
Detectable after P30 |
| rd10 | Rod-specific PDE (β subunit) MISSENSE
Autosomal recessive |
rod-cone
center-to-periphery gradient |
P18 | P14-P28 | P25- | P25-
(regression of fully developed dendrites) |
YES
(data available from P45 to 7 months) |
Best detected after P60 |
Acknowledgments
The authors thank Drs. N. Hawes and B. Chang for the gift of founder rd10 mice, Prof. L. Cervetto and Dr. L. Galli-Resta for critically reading the manuscript, Drs. A. Asta and E. Novelli for technical support, GC. Cappagli and C. Orsini for computer assistance. Funded by the CNR, the NIH grant R01-EY12654 to ES, the MIUR 2006 to CG.
References
- Acland GM, Aguirre GD, Ray J, Zhang Q, Aleman TS, Cideciyan AV, Pearce-Kelling SE, Anand V, Zeng Y, Maguire AM, Jacobson SG, Hauswirth WW, Bennett J. Gene therapy restores vision in a canine model of childhood blindness. Nat Genet. 2001;28:92–95. doi: 10.1038/ng0501-92. [DOI] [PubMed] [Google Scholar]
- Bennett J. Gene therapy for retinitis pigmentosa. Curr Op Mol Ther. 2000;2:420–425. [PubMed] [Google Scholar]
- Bowes C, Danciger M, Kozak CA, Farber DB. Isolation of a candidate cDNA for the gene causing retinal degeneration in the rd mouse. Proc Natl Acad Sci U S A. 1989;86:9722–6. doi: 10.1073/pnas.86.24.9722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowes C, Li T, Danciger M, Baxter LC, Applebury ML, Farber DB. Retinal degeneration in the rd mouse is caused by a defect in the beta subunit of rod cGMP-phosphodiesterase. Nature. 1990;347:677–80. doi: 10.1038/347677a0. [DOI] [PubMed] [Google Scholar]
- Breton ME, Schueller AW, Lamb TD, Pugh EN., Jr Analysis of ERG a-wave amplification and kinetics in terms of the G-protein cascade of phototransduction. Invest Ophthalmol Vis Sci. 1994;35:295–309. [PubMed] [Google Scholar]
- Casini G, Brecha NC, Bosco L, Rickman DW. Developmental expression of Neurokinin-1 and neurokinin-3 receptors in the rat retina. J Comp Neurol. 2000;421(2):275–87. [PubMed] [Google Scholar]
- Chader GJ. Animal models in research on retinal degenerations: past progress and future hope. Vision Res. 2002;42:393–399. doi: 10.1016/s0042-6989(01)00212-7. [DOI] [PubMed] [Google Scholar]
- Chang GQ, Hao Y, Wong F. Apoptosis: final common pathway of photoreceptor death in rd, rds, and rhodopsin mutant mice. Neuron. 1993;11:595–605. doi: 10.1016/0896-6273(93)90072-y. [DOI] [PubMed] [Google Scholar]
- Chang B, Hawes NL, Hurd RE, Davisson MT, Nusinowitz S, Heckenlively JR. Retinal degeneration mutants in the mouse. Vision Res. 2002;42:517–525. doi: 10.1016/s0042-6989(01)00146-8. [DOI] [PubMed] [Google Scholar]
- Cuenca N, Pinilla I, Sauve Y, Lu B, Wang S, Lund RD. Regressive and reactive changes in the connectivity patterns of rod and cone pathways of P23H transgenic rat retina. Neuroscience. 2004;127:301–317. doi: 10.1016/j.neuroscience.2004.04.042. [DOI] [PubMed] [Google Scholar]
- Demontis GC, Sbrana S, Gargini C, Cervetto L. A simple and inexpensive light source for research in visual neuroscience. J Neurosci Meth. 2005;146:13–21. doi: 10.1016/j.jneumeth.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Dhingra A, Lyubarsky A, Jiang M, Pugh EN, Jr, Birnbaumer L, Sterling P, Vardi N. The light response of ON bipolar neurons requires G[alpha]o. J Neurosci. 2000;20:9053–8. doi: 10.1523/JNEUROSCI.20-24-09053.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farber DB, Flannery JG, Bowes-Richman C. The rd mouse: 70 years of research on an animal model of inherited retinal degeneration. Prog Ret Eye Res. 1994;13:31–64. [Google Scholar]
- Fleischhauer J, Njoh WA, Niemeyer G. Syndromic retinitis pigmentosa: ERG and phenotypic changes. Klin Monatsbl Augenheilkd. 2005;222(3):186–90. doi: 10.1055/s-2005-857973. [DOI] [PubMed] [Google Scholar]
- Fox DA, Poblenz AT, He L, Harris JB, Mediano CJ. Pharmacological strategies to block photoreceptor apoptosis caused by calcium overload: a mechanistic target-site approach to neuroprotection. Eur J Ophthalmol. 2003;13 Suppl 3:S44–56. doi: 10.1177/112067210301303s08. [DOI] [PubMed] [Google Scholar]
- Gargini C, Bisti S, Demontis GC, Valter K, Stone J, Cervetto L. ERG changes associated with retinal upregulation of trophic factors: observations following optic nerve section. Neuroscience. 2004;126:775–83. doi: 10.1016/j.neuroscience.2004.04.028. [DOI] [PubMed] [Google Scholar]
- Gargini C, Demontis GC, Cervetto L, Bisti S. Analysis of pharmacologically isolated components of the ERG. Vision Res. 1999;39:1759–1766. doi: 10.1016/s0042-6989(98)00281-8. [DOI] [PubMed] [Google Scholar]
- Ghosh KK, Bujan S, Haverkamp S, Feigenspan A, Wässle H. Types of bipolar cells in the mouse retina. J Comp Neurol. 2004;26;469(1):70–82. doi: 10.1002/cne.10985. [DOI] [PubMed] [Google Scholar]
- Guillery RW. On counting and counting errors. J Comp Neurol. 2002;447(1):1–7. doi: 10.1002/cne.10221. [DOI] [PubMed] [Google Scholar]
- Hart AW, McKie L, Morgan JE, Gautier P, West K, Jackson IJ, Cross SH. Genotype-phenotype correlation of mouse pde6b mutations. Invest Ophthalmol Vis Sci. 2005;46:3443–3450. doi: 10.1167/iovs.05-0254. [DOI] [PubMed] [Google Scholar]
- Haverkamp S, Ghosh KK, Hirano AA, Wassle H. Immunocytochemical description of five bipolar cell types of the mouse retina. J Comp Neurol. 2003;455(4):463–76. doi: 10.1002/cne.10491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BW, Watt CB, Frederick JM, Baehr W, Chen CK, Levine EM, Milam AH, Lavail MM, Marc RE. Retinal remodeling triggered by photoreceptor degenerations. J Comp Neurol. 2003;464:1–16. doi: 10.1002/cne.10703. [DOI] [PubMed] [Google Scholar]
- Lamb TD, Pugh EN., Jr A quantitative account of the activation steps involved in phototrasduction in amphibian photoreceptors. J Physiol. 1992;449:719–758. doi: 10.1113/jphysiol.1992.sp019111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVail MM, Matthes MT, Yasumura D, Steinberg RH. Variability in rate of cone degeneration in the retinal degeneration (rd/rd)mouse. Exp Eye Res. 1997;65:45–50. doi: 10.1006/exer.1997.0308. [DOI] [PubMed] [Google Scholar]
- Leveillard T, Mohand-Said S, Lorentz O, Hicks D, Fintz AC, Clerin E, Simonutti M, Forster V, Cavusoglu N, Chalmel F, Dolle P, Poch O, Lambrou G, Sahel JA. Identification and characterization of rod-derived cone viability factor. Nat Genet. 2004;36:755–9. doi: 10.1038/ng1386. [DOI] [PubMed] [Google Scholar]
- Loewenstein JI, Montezuma SR, Rizzo JF., 3rd Outer retinal degeneration: an electronic retinal prosthesis as a treatment strategy. Arch Ophthalmol. 2004;122(4):587–96. doi: 10.1001/archopht.122.4.587. [DOI] [PubMed] [Google Scholar]
- Lund RD, Coffey PJ, Sauve Y, Lawrence JM. Intraretinal transplantation to prevent photoreceptor degeneration. Ophthalm Res. 1997;29:305–319. doi: 10.1159/000268029. [DOI] [PubMed] [Google Scholar]
- Lyubarsky AL, Daniele LL, Pugh EN., Jr From candelas to photoisomerizations in the mouse eye by rhodopsin bleaching in situ and the light-rearing dependence of the major components of the mouse ERG. Vision Res. 2004;44:3235–3251. doi: 10.1016/j.visres.2004.09.019. [DOI] [PubMed] [Google Scholar]
- Lyubarsky AL, Pugh EN., Jr Recovery phase of the murine rod photoresponse reconstructed from electroretinographic recording. J Neurosci. 1996;16:563– 571. doi: 10.1523/JNEUROSCI.16-02-00563.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marc RE, Jones BW, Watt CB, Strettoi E. Neural remodeling in retinal degeneration. Prog Retin Eye Res. 2003;22:607–655. doi: 10.1016/s1350-9462(03)00039-9. [DOI] [PubMed] [Google Scholar]
- McKernan DP, Caplis C, Donovan M, O’brien CJ, Cotter TG. Age-dependent susceptibility of the retinal ganglion cell layer to cell death. Invest Ophthalmol Vis Sci. 2006;47(3):807–14. doi: 10.1167/iovs.05-0520. [DOI] [PubMed] [Google Scholar]
- Nomura A, Shigemoto R, Nakamura Y, Okamoto N, Mizuno N, Nakanishi S. Developmentally regulated postsynaptic localization of a metabotropic glutamate receptor in rat rod bipolar cells. Cell. 1994;77(3):361–9. doi: 10.1016/0092-8674(94)90151-1. [DOI] [PubMed] [Google Scholar]
- Ogilvie JM, Tenkova T, Lett JM, Speck J, Landgraf M, Silverman MS. Age-related distribution of cones and ON-bipolar cells in the rd mouse retina. Curr Eye Res. 1997;16:244–51. doi: 10.1076/ceyr.16.3.244.15411. [DOI] [PubMed] [Google Scholar]
- Otani A, Dorrell MI, Kinder K, Moreno SK, Nusinowitz S, Banin E, Heckenlively J, Friedlander M. Rescue of retinal degeneration by intravitreally injected adult bone marrow-derived lineage-negative hematopoietic stem cells. J Clin Invest. 2004;114:765–774. doi: 10.1172/JCI21686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce EA. Pathways to photoreceptor cell death in inherited retinal degenerations. Bioessays. 2001;23:605–18. doi: 10.1002/bies.1086. [DOI] [PubMed] [Google Scholar]
- Peichl L, Gonzalez-Soriano J. Morphological types of horizontal cell in rodent retinae: a comparison of rat, mouse, gerbil, and guinea pig. Vis Neurosci. 1994;11(3):501–17. doi: 10.1017/s095252380000242x. [DOI] [PubMed] [Google Scholar]
- Pignatelli V, Cepko CL, Strettoi E. Inner retinal abnormalities in a mouse model of Leber’s congenital amaurosis. J Comp Neurol. 2004;469:351–359. doi: 10.1002/cne.11019. [DOI] [PubMed] [Google Scholar]
- Pignatelli V, Strettoi E. Bipolar cells of the mouse retina: a gene gun, morphological study. J Comp Neurol. 2004;476:254–66. doi: 10.1002/cne.20207. [DOI] [PubMed] [Google Scholar]
- Pugh EN, Jr, Lamb TD. Amplification and kinetics of the activation steps in phototransduction. Biochim Biophys Acta. 1993;1141:111–149. doi: 10.1016/0005-2728(93)90038-h. [DOI] [PubMed] [Google Scholar]
- Remè CE, Grimm C, Hafezi F, Marti A, Wenzel A. Apoptotic cell death in retinal degenerations. Prog Ret Eye Res. 1998;17:443–464. doi: 10.1016/s1350-9462(98)00009-3. [DOI] [PubMed] [Google Scholar]
- Rex TS, Allocca M, Domenici L, Surace EM, Maguire AM, Lyubarsky A, Cellerino A, Bennett J, Auricchio A. Systemic but not intraocular Epo gene transfer protects the retina from light-and genetic-induced degeneration. Mol Ther. 2004;10:855–861. doi: 10.1016/j.ymthe.2004.07.027. [DOI] [PubMed] [Google Scholar]
- Saszik SM, Robson JG, Frishman LJ. The scotopic threshold response of the dark-adapted electroretinogram of the mouse. J Physiol. 2002;543:899–916. doi: 10.1113/jphysiol.2002.019703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmued LC, Albertson C, Slikker W., Jr Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 1997;751:37–46. doi: 10.1016/s0006-8993(96)01387-x. [DOI] [PubMed] [Google Scholar]
- Sharma RK, O’Leary TE, Fields CM, Johnson DA. Development of the outer retina in the mouse. Brain Res Dev Brain Res. 2003;145:93–105. doi: 10.1016/s0165-3806(03)00217-7. [DOI] [PubMed] [Google Scholar]
- Strettoi E, Pignatelli V, Hageman GS. Morphological abnormalities of bipolar cells in human retinits pigmentosa. Soc for Neurosci Abstr. 2005:977.2. [Google Scholar]
- Strettoi E, Porciatti V, Falsini B, Pignatelli V, Rossi C. Morphological and functional abnormalities in the inner retina of the rd/rd mouse. J Neurosci. 2002;22:5492–5504. doi: 10.1523/JNEUROSCI.22-13-05492.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardi N, Dhingra A, Zhang L, Lyubarsky A, Wang TL, Morigiwa K. Neurochemical organization of the first visual synapse. Keio J Med. 2002;51:154–64. doi: 10.2302/kjm.51.154. [DOI] [PubMed] [Google Scholar]
- Varela C, Igartua I, De la Rosa EJ, De la Villa P. Functional modifications in rod bipolar cells in a mouse model of retinitis pigmentosa. Vision Res. 2003;43:879–85. doi: 10.1016/s0042-6989(02)00493-5. [DOI] [PubMed] [Google Scholar]
- Wang S, Lu B, Lund RD. Morphological changes in the Royal College of Surgeons rat retina during photoreceptor degeneration and after cell-based therapy. J Comp Neurol. 2005;491:400–17. doi: 10.1002/cne.20695. [DOI] [PubMed] [Google Scholar]
- Zeiss CJ, Neal J, Johnson EA. Caspase-3 in postnatal retinal development and degeneration (2004) Invest Opht]halmol Vis Sci. 45:964–70. doi: 10.1167/iovs.03-0439. [DOI] [PubMed] [Google Scholar]