

Abstract
Dysregulated activity of phosphatidylinositol 3-kinase (PI3K) and mammalian target of rapamycin complex 1 (mTORC1) is characteristic feature of hamartoma syndromes. Hamartoma syndromes, dominantly inherited cancer predisposition disorders, affect multiple organs and are manifested by benign tumors consisting of various cell types native to the tissues in which they arise. In the past few years, three inherited hamartoma syndromes, Cowden syndrome (CS), tuberous sclerosis complex (TSC) syndrome, and Peutz-Jeghens syndrome (PJS), have all been linked to a common biochemical pathway: the hyperactivation of PI3K/mTORC1 intracellular signaling. Three tumor suppressors, PTEN (phosphatases and tensin homolog), tuberous sclerosis complex TSC1/TSC2, and LKB1, are negative regulators of PI3K/mTORC1 signaling; disease-related inactivation of these tumor suppressors results in the development of PTEN-associated hamartoma syndromes, TSC and PJS, respectively. The goal of this review is to provide a roadmap for navigating the inherently complex regulation of PI3K/mTORC1 signaling while highlighting the progress that has been made in elucidating the cellular and molecular mechanisms of hamartoma syndromes and identificating potential therapeutic targets for their treatment. Importantly, because the PI3K/mTORC1 pathway is activated in the majority of common human cancers, the identification of novel molecular target(s) for the treatment of hamartoma syndromes may have a broader translational potential, and is critically important not only for therapeutic intervention in hamartoma disorders, but also for the treatment of cancers.
Keywords: PI3K, mTORC1, PTEN, cancer, lung, kidney, TSC, LAM, biomarker, therapy
Linking Gene Mutations to Hamartoma Syndromes
Hamartoma syndromes are manifested by benign tumors in multiple organs, and represent a tumorous mass with anomalous development of differentiated cells resembling normal cells to normal tissue rather then true tumor. For example, pulmonary lymphangioleiomyomatosis (LAM),1 rare lung disease, which could be sporadic or strikes women with tuberous sclerosis complex (TSC) hamartoma syndrome,2,3 manifests by abnormal growth of smooth muscle-like cells forming nodules, which leads to the cystic destruction of the lungs and pneumothorax.4,5 Although hamartomas are benign tumors, affected individuals have an increased risk of developing certain malignant cancers, and they usually also have a decreased life span compared to the general population.
For the last 10 years, the short history of hamartoma syndromes includes significant breakthroughs, from the identification of genes mutated in these diseases to the discovery of the functions of these genes. These findings provided the molecular basis and foundation for the development of potential therapeutic strategies for treatment of some hamartoma syndromes.6–9 Thus, mutational inactivation of tumor suppressor proteins tuberous sclerosis complex 1 (TSC1)/TSC2, LKB1 and PTEN (phosphatases and tensin homolog) are linked, respectively, to tumor development and pathophysiology in hamartoma syndromes TSC,10–12 Peutz-Jeghens syndrome (PJS),13,14 and the PTEN-associated hamartoma syndromes including Cowden syndrome (CS),15,16 Bannayan-Riley-Ruvalcaba syndrome (BRRS),17 Proteus syndrome (PS),18 and Proteus-like syndrome (PLS).19–21 The diversity of disease manifestations in hamartoma syndromes is apparent; however, they all share one common feature that is the dysregulation of the mTORC1 signaling pathway. The mechanism by which mTORC1 is regulated by tumor suppressor complex TSC1/TSC2, LKB1 and PTEN signaling led to a growing appreciation that mTORC1 mediates three major inputs from growth factors, amino acids and ATP, and is a master regulator of cell growth and metabolism. In normal cells, mTORC1 signaling is tightly controlled; however, in hamartoma syndromes and in cancer, mutations of the pathway, which constitute one of the most prevalent classes of tumor-associated mutations in humans, result in neoplastic growth.22 Tumor suppressors PTEN and TSC1/TSC2 belong to the phosphatidylinositol 3-kinase (PI3K) signaling cascade, a highly connected and conserved signal transduction network activated by growth-promoting signals that regulate cell proliferation and survival (Fig. 1).23–26 Tumor suppressor LKB1 regulates mTORC1 signaling by providing an energy-sensing input into the PI3K signaling cascade on the level of TSC1/ TSC2 (Fig. 1).27
Figure 1.

mTORC1 signaling. mTORC1 is a master regulator of cell growth and metabolism, which mediates three major inputs from growth promoting signals such, for example, growth factors and insulin. mTORC1 directly phosphorylates and activates S6K1 and inhibit 4E-BP1, which leads to protein synthesis, cell growth and proliferation. mTORC1 is positively regulated by Akt-PRAS40 and negatively regulated by tumor suppressor complex TSC1/TSC2, which suppress activity of small GTPase Rheb. Rheb activity is positively regulated by mTCTP. TSC1/TSC2 activity is suppressed by inhibitory phosphorylation of TSC2 by PI3K-dependent Akt, ERK1/2 and ERK1/2-dependent RSK1. PI3K is activated by multiple inputs such as growth factors and insulin, which leads to recruitment of PI3K to the membrane and its binding IRS-1. Active PI3K converts phosphatidylinositol-4,5-biphosphate (PIP2) to phosphatidylinositol-3,4,5-trisphosphate (PIP3), which leads to PDK1-dependent phosphorylation of Akt followed by activation of mTOR in the mTORC1 complex. mTOR is part of two distinct complexes: the rapamycin-sensitive mTORC1 consisting of mTOR, raptor and adaptor protein GβL, an upstream activator of S6K1, and the rapamycin-insensitive mTORC2, consisting of mTOR, rictor and GβL. mTORC2 directly phosphorylates Akt, which is necessary for full Akt activation. In contrast, hyperactivated S6K1 inhibits IRS1-PI3K-Akt signaling pathway (the feedback loop). Tumor suppressor PTEN antagonizes PI3K-Akt signaling by specifically dephosphorylating PIP3 to PIP2. TSC1/TSC2 activity is positively regulated by cellular energy: increase in AMP levels due to glucose or fatty acids depravation and other stresses leads to LKB1-dependent activation of AMPK; active AMPK directly phosphorylates TSC2 and primes TSC2 for its subsequent phosphorylation by GSK3, which leads to inhibition of mTORC1, inhibition of translation, protein synthesis and cell growth. Pink or blue coloring indicate signaling molecules which are either positively or negatively, respectively, regulate cell growth and proliferation. Arrows indicate functional enhancement; flat bars indicate functional suppression.
The goal of this review is to provide a roadmap for navigating the inherent complexity and regulation of the PI3K/ mTORC1 signaling pathway while pointing out potential therapeutic targets to treat hamartoma syndromes; we will also provide a list of references of original findings and substantial reviews done on mTORC1 signaling. Because the activity of TSC1/ TSC2 is regulated by PI3K and PTEN counteracts the effects of PI3K, we will start our review with a brief description of PI3K signaling. Additionally, because PI3K activity is influenced by prolonged treatment with mTORC1 inhibitor rapamycin, we will also review current advances in therapeutic targeting of PI3K28,29 that might be beneficial for the combination therapy of hamartoma syndromes.
PI3K
PI3Ks are a family of lipid kinases that catalyze the addition of phosphate molecule specifically to the 3′-OH position of the inositol ring of phosphoinositides converting phosphatidylinositol-4,5-biphosphate (PIP2) to phosphatidylinositol-3,4,5-trisphosphate (PIP3) at the membrane.30–33 PI3K is activated by multiple inputs such as growth factors, insulin, cytokines, cell-cell and cell-matrix interactions provided by both receptor and non-receptor tyrosine kinases and adhesion molecules.34 PI3K isoforms can be divided into three classes based on their structure and substrate specificity.35,36 Class IA PI3Ks are cytoplasmic heterodimers composed of a 110-kDa (p110α, β or δ) catalytic subunit and an 85-kDa (p85, p55 or p50) adaptor protein. Catalytic subunits p110α and p110β are ubiquitously expressed in mammalian cells. Catalytic subunit p110δ is expressed predominantly in lymphocytes and lymphoid tissues and therefore, may play a role in PI3K-mediated signaling of immune responses.37 Class IA isoforms are mainly activated by receptor and nonreceptor tyrosine kinases while class IB p110γ, first identified during the screening of human bone marrow cDNA, is activated by Gβγ subunits of G protein coupled receptors (GPCRs)38 and by T cell receptor tyrosine kinase activated protein.39 Class II isoforms are mainly associated with the phospholipid membranes and are present in the endoplasmic reticulum and Golgi apparatus.40 Class III isoforms, structurally related to yeast vesicular sorting protein Vps34p,41 are mammalian homologues that use only membrane phosphatidylinositol as a substrate and generate phosphatidylinositol-3-monophosphate.
Identification of PI3K isoforms and elucidation of their cellular functions revealed the critical role of PI3K signaling in normal cell functions; meanwhile, growing evidence also linked dysregulation of PI3K signaling to cancer. Mutations in the PI3KCA gene, which is located in common amplicon at 3q26 and encodes the p110α, catalytic subunit of PI3K, were found in more then 30% of various solid tumor types including ovary, cervix and head and neck cancers.42 Functional analysis of these mutations showed that they increased PI3K enzymatic activity and stimulated Akt signaling thus allowing growth factor-independent growth as well as increased cell migration and metastasis.35 Somatic mutations in the PI3KR1 gene encoding the p85α regulatory subunit of PI3K leading to the PI3K constitutive activation, had been detected in human ovarian and colon tumors and human lymphoma cells.43,44 While mutation of PI3Ks has been predominantly associated with different types of cancers,22,36 specific PI3K mutations had not been linked to hamartoma syndromes. However, in hamartoma syndromes PI3K signaling is activated due to PTEN inactivating mutations, which makes PI3K a critical target for potential therapeutic intervention in hamartoma syndromes, which will be discussed below.
mTORC1
The relevance of mTORC1 in hamartoma syndromes arises from the discovery of the constitutive activation of this canonical pathway in pulmonary LAM12 and TSC45,46 due to the inactivating mutations of tumor suppressor complex TSC1/TSC2. Because mTORC1 is a downstream effector of PI3K and forms a canonical PI3K-mTORC1 signaling cascade, these findings also underscore the impact that mTORC1 over-activation has on the PTEN-associated hamartoma syndromes and identified mTORC1 as a molecular target for cancer treatment. mTOR, a highly conserved serine-threonine kinase, was identified as a molecular target of rapamycin, a macrolide extracted from the fungi Streptomyces hydroscopicus.47 Rapamycin forms a cytosolic complex with FK506 binding protein 12 (FKBP12), which inhibits the catalytic activity of mTOR.48,49 Specific inhibition of mTORC1 activity by rapamycin and the fact that mTORC1 is constitutively activated not only in hamartoma syndromes but in many different types of cancer, led to multiple rapamycin clinical trials; results of some of these trials have already been published6,50,51 and are discussed in this review.
mTORC1 consists of the serine-threonine kinase mTOR, regulatory-associated protein of mTOR (raptor),52,53 mLST8 (also known as GβL)54 and the proline-rich Akt substrate of 40 kDa (PRAS40) (Fig. 1). In mTORC1, mTOR provides mTORC1 the catalytic activity, and raptor functions as a positive regulator of mTOR activity and expression.52,53,55 The activity of raptor towards mTOR can be regulated by p90 ribosomal protein S6 kinase-dependent phosphorylation56 and energy-stress conditions through AMP-activated protein kinase (AMPK).57 PRAS40 acts as an inhibitor of mTOR kinase activity in an Akt-dependent phosphorylation manner.58–60 Interestingly, some studies have demonstrate that PRAS40 can also be phosphorylated by activated mTOR at Akt-independent sites, which promotes the release of PRAS40 from mTORC1 and increases mTORC1 activity.61,62 The function of mLST8 in mTORC1 remains to be established.63
Active mTORC1 directly phosphorylates S6K1 and 4E-BP1, which leads to SK61 activation and 4E-BP1 inhibition followed by protein translation, ribosome biogenesis and cell cycle progression.64,65 mTORC1 mediates inputs from three major classes of molecules: growth factors, amino acids and ATP, and is a master regulator of cell growth and metabolism.66 In normal cells, mTORC1 signaling is tightly controlled; however, in hamartoma syndromes and in cancer, mutations of the pathway result in benign and neoplastic growth.22
mTORC1 is negatively regulated by tumor suppressor complex TSC1/TSC2. TSC2 functions as a GTPase activating protein (GAP) for the small GTPase Rheb (Ras-related small G protein Ras homologue enriched in brain). Growth factor- or insulin-induced activation of PI3K, which converts PIP2 to PIP3 at the membrane,30–33 leads to Akt activation which, in turn, leads to the Akt-dependent inactivating phosphorylation of TSC2. It inhibits TSC2 Rheb GAP activity followed by the activation of Rheb, which directly binds to and activates mTORC1 (Fig. 1).67–71 Insulin-induced activation of Akt, in addition to TSC1/TSC2 inhibition, also leads to the direct phosphorylation of PRAS40, which is thus unbound from mTORC1 and increases mTORC1 activity.58–60 In addition to FKBP12, another member of the FK506-binding protein family, FKBP38, a mitochondrial membrane protein, was identified as an inhibitor of mTORC1 activation.72 Apparently, in a quiescent state, FKBP38 is bound to mTOR; then, upon cell growth signaling, GTP-bound small GTPase Rheb (Fig. 1) binds to FKBP38 and induces the release and activation of mTOR.73 FKBP38 binds to mTOR and inhibits its activity in a manner similar to that of the FKBP12-rapamycin complex. Interestingly, FKBP38 binds to a region of mTOR that apparently overlaps with another mTOR region that binds the FKBP12-rapamycin complex, suggesting that the later may preclude binding of FKBP38 to mTOR.73 However, the precise role of FKBP38 in the canonical mTORC1 signaling pathway remains to be established. The activity of mTOR can also be regulated by targeted ubiquitination and consequent degradation by binding to the novel tumor suppressor protein F-box and WD repeat domain containing 7 (FBXW7).74 Interestingly, tumor cells with loss or mutation of FBXW7 are particularly sensitive to rapamycin treatment.74
In addition to the canonical rapamycin-sensitive mTORC1 signaling, mTOR also forms a physically and functionally distinct complex—the rapamycin-insensitive mTORC2,75 consisting of mTOR, rictor,75 GβL76 and mSIN1 (mammalian stress-activated protein kinase-interaction protein 1).77 In vivo78 and in vitro79 studies established the role of mTORC2 as the long-sought phosphoinositide-dependent kinase 2 (PDK2), which phosphorylates Akt at Ser473 (Fig. 1).79
TSC1/TSC2
Tumor suppressors TSC1 and TSC2 form a physical and functional complex, in which TSC1 functions as the regulatory component stabilizing TSC2, and facilitating the TSC2 catalytic function as a GAP for the small GTPase Rheb, a negative regulator of mTORC1. The role of TSC1/TSC2 in regulating the mTORC1 signaling pathway has been intensively studied, and it has been clearly demonstrated that the TSC1/TSC2 complex is a key regulator of mTORC1 signaling. Comprehensive reviews of the manner in which dysregulation of TSC1/TSC2-mTORC1 modulates intracellular signaling, for example, the negative feedback inhibition of insulin signaling and Akt activation, are published in refs.5,21,80,81
TSC1/TSC2 activity is regulated by the PI3K signaling cascade, a highly connected and conserved signal transduction network activated by growth-promoting signals that regulate cell proliferation and survival.23–26 PI3K-Akt regulates the activity of TSC1/TSC2 due to Akt-dependent TSC2 phosphorylation at multiple sites (Fig. 1).5 Interestingly, while the multiple Akt-dependent sites that inactivate TSC2 Rheb GAP activity leading to mTORC1 activation have been identified, the physiological significance of Akt-dependent TSC2 phosphorylation have not been completely elucidated. Conflicting evidence comes from studies in Drosophila showing that mutation of Akt phosphorylation sites on TSC2 neither prevented S6 activation by insulin nor affected Drosophila development.82 Rheb activity is also positively regulated by the guanine nucleotide exchange factor activity of translationally controlled tumor protein (TCTP);83 TCTP appears to act downstream or parallel to TSC1/TSC2. The precise role of TCTP in growth-promoting activation of Rheb and mTORC1 in mammalians remains to be elucidated.84
TSC1/TSC2 activity is also negatively regulated by extracellular signal-regulated kinases (ERK). ERK-dependent TSC2 phosphorylation leads to dissociation of the TSC1 and TSC2 complex, which results in mTOR/S6K1 activation, increased cell proliferation and transformation.85
In addition to the growth-promoting signaling mediated by PI3K-Akt and ERK, TSC1/TSC2 activity is regulated by cellular energy, that is, the availability of glucose and ATP. Energy starvation, including glucose deprivation and other stresses, such as attenuation of ATP synthesis during hypoxia or increased ATP consumption during physical exertion, results in the activation of AMPK by an upstream kinase LKB1.86,87 Activated AMPK directly phosphorylates TSC2; this is then followed by inhibition of mTORC1 leading to inhibition of translation and attenuation of protein synthesis.88,89 Recent data, however, indicates that, under energy stress conditions, AMPK may regulate mTORC1 activity in a TSC2-independent manner via the direct phosphorylation of raptor, which leads to mTORC1 inhibition and cell cycle arrest.90 Additionally, genotoxic stress negatively regulates TSC1/TSC2-mTORC1 signaling in a p53-AMPK-dependent manner.91 Genotoxic stress activation of p53 leads to the expression of sestrin1 and sestrin2, two novel proteins, which activate AMPK leading to AMPK-dependent inhibiting phosphorylation of TSC2 thereby inhibiting mTORC1 and consequently cell growth and proliferation.92
AMPK phosphorylation primes TSC2 for its subsequent phosphorylation by glycogen synthase kinase 3 (GSK3).93 GSK3 activity is negatively regulated by the Wnt signaling pathway, which plays a critical role in cellular proliferation, especially during development.57 Wnt-dependent activation of S6K1 requires the inhibition of GSK3 and TSC1/TSC2 activity.93 Inhibition of GSK3 activity by Wnt is specific and distinct from its inhibition by Akt and RSK1.94 Thus, TSC2 integrates growth, energy and Wnt signaling in coordinated inhibition of mTORC1 signaling, protein translation and cell growth.
Mutational inactivation of tumor suppressor proteins TSC1 or TSC2 are linked, respectively, to tumor development and the pathophysiology in hamartoma syndrome TSC.5,10–12 Positional cloning and linkage analysis in multigenerational families with TSC were used to map both TSC1 and TSC2 genes.10,11 Patients with TSC develop hamartomas and benign tumors in the brain, heart and kidney; they can exhibit cognitive defects, epilepsy and autism.3,95
At the same time, another disease, pulmonary lymphangioleiomyomatosis (LAM), that could be sporadic or manifested in association with TSC, is also linked to the TSC1 and TSC2 genes.4,5,12 Patients with LAM develop lung tumors characterized by abnormal smooth muscle-like cell growth, cystic destruction of the lungs, and loss of pulmonary function.1,2 Extensive genetic analysis of TSC1 or TSC2 genes in TSC revealed a wide spectrum of mutations; however, no particular region was linked to higher mutation rate.3 Importantly, mutations of TSC2 account for more than 60% of the cases, and are associated with the severe clinical TSC phenotype.3,5 Loss of function mutations or loss of heterozygosity (LOH) of either TSC1 or TSC2 lead to the constitutive activation of the mTORC1 signaling pathway and abnormal cell growth in TSC hamartoma syndrome and pulmonary LAM.5,12,96,97
In addition to constitutive mTORC1-S6K1 activation, TSC1/ TSC2 loss of function causes the deregulation of other mTORC1-dependent and independent signaling pathways. It was shown that TSC2-related upregulation of mTORC1 activity causes endoplasmic reticulum stress in TSC2-deficient cell and tumor models98 and deregulated glucose transporter Glut4 trafficking.99 Additionally, expression of the platelet-derived growth factor (PDGF) receptor, one of the targets of negative feedback regulation in cells with activated mTORC1, is suppressed in TSC2-null cells.100 Furthermore, constitutive mTORC1 activation due to loss of TSC2 amplifies p53 activation by stimulating p53 translation, which results in the increased accumulation of p53 and apoptosis in response to stress conditions.101 Importantly, loss of TSC2 function also leads to mTORC1-independent constitutive activation of the small GTPase RhoA, a well known positive regulator of cell proliferation and motility, in LAM102 and TSC2-null ELT3 cells.103,104 While additional studies are needed to establish the contribution of ER stress and Rho GTPase in the mTORC1-related hamartomas, it is possible that some of these TSC1/TSC2-related signaling abnormalities may contribute to benign tumor formation.
PTEN
Tumor suppressor PTEN negatively regulates PI3K-mTORC1 signaling by specifically dephosphorylating the 3′-OH position of the inositol ring of the lipid second messengers PIP3 (Fig. 1).105–107 Loss of tumor suppressor PTEN leads to upregulation of the PI3K signaling pathway which results in the elevation of PIP3 levels and contributes to oncogenesis (Fig. 2).108
Figure 2.
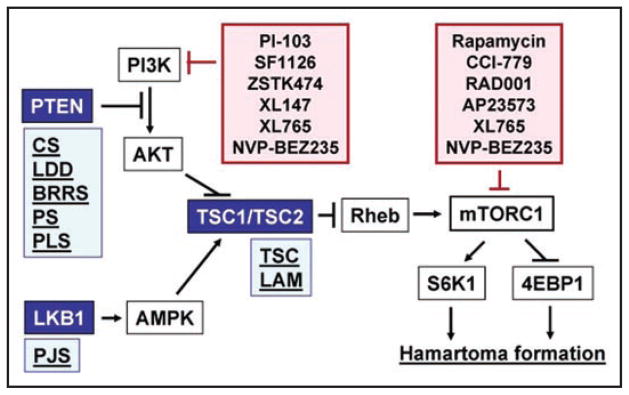
Potential targeting of PI3K/mTORC1 signaling in hamartoma syndromes. Germline mutations of PTEN, LKB1, TSC1 and TSC2 genes lead to upregulation of PI3K/mTORC1 signaling and are associated with spectrum of hamartoma syndromes (Light blue boxes). Blue coloring indicate tumor suppressor proteins, negatively regulating mTORC1 signaling. Germline mutations of PTEN is associated with Cowden syndrome (CS), Lhermitte-Duclos disease (LDD), Bannayan-Riley-Ruvalcaba syndrome (BRRS), Proteus syndrome (PS), and Proteus-Like syndrome; germline mutations of LKB1 is associated with Peutz-Jeghers syndrome (PJS), TSC1 or TSC2 mutations is associated with Tuberous Sclerosis Complex (TSC) and Lymphangioleiomyomatosis (LAM). Pink coloring indicate inhibitors of PI3K/ mTORC1 signaling pathway, which may have promising therapeutic effects for treatment of hamartoma syndromes. Rapamycin and it analogs CCI-779, RAD001 and AP23573 are to inhibit mTORC1; PI-103, SF11206 and ZSTK474 are pan-PI3K inhibitors; NVP-BEZ235 and XL765 simultaniously target mTORC1 and PI3K.
The germline inactivating mutations of PTEN are associated with a predisposition to autosomal-dominant hamartoma syndromes42,109,110 with different phenotypic features including Cowden syndrome (CS),15,16 Bannayan-Riley-Ruvalcaba syndrome (BRRS),17 Proteus syndrome (PS),18 and Proteus-like syndrome (PLS).19,111 In over 85% of cases, CS results from germline PTEN mutations in promoter region, and are characterized by multiple hamartomas of the hair follicle, the mucocutaneous membranes, breast, thyroid and intestinal tissues, and an increased risk of breast, thyroid and endometrial cancer.42 The characteristic features of BRRS are lipomatosis, macrocephaly, high birth weight, and in males, speckled penis and no association with an increased risk of malignancy.109,112 Germline PTEN mutations such as deletion of 10q23 and translocation involving 10q23 has been found in more then 60% of BRRS patients.111 Interestingly, that mutational spectra of BRRS and CS overlaps. PS is a disorder of patchy or mosaic postnatal overgrowth of unknown etiology such as asymmetric limb overgrowth and length discrepancy, macrodactyly, vertebral abnormalities, hyperostosis, abnormal and asymmetric fat distribution, asymmetric muscle development, connective-tissue nevi and vascular malformations such as deep venous thrombosis and pulmonary embolism.113 Because of mosaic pattern of distribution, patients with PS manifest variable phenotype. The germline PTEN mutations have been detected in 10–20% cases of PS.111 PLS is diagnosed in patients with overgrowth which does not meet established diagnostic criteria for PS.111 60% patients with PLS carry germline PTEN mutations111 including R335X and mosaic R130X.19
LKB1
The tumor suppressor LKB1, also known as STK11 (serine-threonine kinase 11), is a serine/threonine kinase that is mutationally inactivated in PJS hamartoma syndrome.13,27 LKB1 serves to activate AMPK by direct phosphorylation of Thr172 in its activation loop, which is essential for AMPK catalytic activity. AMPK, a cellular energy sensor, functions as a master regulator of cellular energy metabolism. Depletion of intracellular ATP levels due to either physiological stimuli such as exercise and muscle contraction or pathological stresses such as hypoxia, oxidative stress and glucose deprivation activate AMPK.87 AMPK functions to restore intracellular ATP levels by inhibiting ATP-consuming processes such as protein translation and cell growth and promoting ATP-generating processes such as gluconeogenesis and lipogenesis.114 Thus, under energy starvation AMPK inhibits cell proliferation by directly phosphorylating TSC2 and enhancing its ability to switch off mTORC1 signaling.115 Mutational inactivation of LKB1 results in hyperactivation of mTORC1 signaling under low energy conditions,116,117 suggesting that LKB1 is required for repression of mTORC1 in a AMPK- and TSC2-dependent manner.
Germline mutations of LKB1 are documented in more than 70–80% of PJS patients; up to 15% of cases have germline deletions of all, or part of LKB1. Hamartoma syndrome in PJS manifests by mucocutaneous pigmentation defects and hamartomatous polyps in the gastrointestinal tract.27 Another key feature of PJS is a high predisposition to the early onset of a variety of carcinoma including small intestine, colorectal, esophageal, pancreatic, breast cancers and adenocarcinoma.118 The majority of patients have a familial history of PJS; however, 10–20% of PJS cases result from spontaneous germline mutations. The majority of disease state due to LKB1 gene mutations results in large truncations of the catalytic domain, which impairs LKB1 catalytic activity. A significant number of point mutations in the kinase domain and in the C-terminal noncatalytic region have been also identified in PJS patients.27
Loss of heterozygocity (LOH), haploinsufficiency and epigenetic inactivation
The prevailing opinion for hamartoma syndromes is that the disease develops in accordance with the classic Knudson’s two-hit tumor suppressor gene model.119 For example, a mutation in either TSC1 or TSC2 is followed by a second hit referred to as loss of heterozygosity (LOH) leading to the loss of function of either TSC1 or TSC2 proteins. LOH in TSC1 or TSC2 has been identified in TSC lesions including angiomyolipomas,3 LAM cells,12 and ependymal giant-cell tumors; however, LOH in TSC1 or TSC2 were rarely found in cerebral cortical tubes, a neurological manifestation of familial TSC.3 Interestingly, in about 15–20% patients with clinical symptoms of TSC, no identifiable mutations in either TSC1 or TSC2 have been found.3 In TSC skin hamartoma, TSC1 or TSC2 two-hit deletions were also not detected in the proliferating epidermal cells.120 In CS-related tumors, hemizygous inactivation of PTEN occurs very frequently, however, loss of the second allele is rare.42 There is the possibility that either LOH or gene mutations were not detected due to the low sensitivity of used technical approaches; or loss of one allele may have a partial tumor promoting effect.42 Importantly, PTEN gene mutations are not the only cause of PTEN inactivation, which can also be due to other mechanisms such as epigenetic inactivation of tumor suppressor genes, transcriptional regulation and posttranslational modifications of tumor suppressor proteins. Thus, epigenetic inactivation of the PTEN was first reported in prostate cancer xenografts due to methylation of PTEN gene promoter and loss of PTEN protein expression.121 Furthermore, nonsense mutations leading to degradation of PTEN mRNA17 or regulation of PTEN levels by TGFβ through p53,122,123 had been linked to the downregulation of PTEN tumor suppressor function. PTEN and TSC2 can also be post-translationally inactivated due to phosphorylation. It was shown that the phosphorylation of the C-terminus of PTEN by CK2,124 regulates protein stability and function.125 ERK-dependent TSC2 phosphorylation leads to dissociation of TSC1 and TSC2 complex, which results in mTOR/ S6K1 activation, increased cell proliferation and transformation.85 Collectively, the evidence demonstrates that hamartoma syndromes can develop not only due to LOH but also due to epigenetic inactivation of tumor suppressor genes, transcriptional regulation and posttranslational modifications of tumor suppressor proteins.
PI3K/mTORC1 Signaling as a Target for the Treatment of Hamartoma Syndromes and the Next Frontier in Drug Discovery
Targeting the mTORC1
There are currently no approved therapies for PHTS, TSC, LAM and PJS in clinic. However, rapamycin, a specific inhibitor of mTORC1, discovered more then 30 years ago, attracts a renewed interest. Rapamycin (sirolimus is the official generic name) is a prototypical inhibitor of mTORC1 signaling.126 It was discovered in early 1970s during the screening for new anti-fungal agents, and was first named rapamycin because it was isolated from a soil sample from island Rapa Nui, the indigenous name of Easter Island in the South Pacific. Rapamycin is a macrocyclic lactone produced by Streptomyces hydroscopicus.126 Rapamycin forms a complex with its intracellular receptor FKBP12 and binds directly to mTORC1, which suppresses mTORC1-mediated signaling.81 However, the precise mechanism of rapamycin-induced inhibition of mTORC1 signaling is not completely understood, and at least two mechanisms of rapamycin action have been proposed. It was shown that under some experimental conditions FKBP12-rapamycin destabilizes the interaction of mTOR with raptor, which is required for mTOR activity.52 Separate studies demonstrate that the FKBP12-rapamycin complex suppresses mTORC1 autophosphorylation, which also may inhibit mTORC1 activity76 suggesting the possibility that the rapamycin-dependent mTORC1 inhibition may involve more then one mechanism.
Because mTORC1 signaling is a highly conserved pathway that regulates protein synthesis and cell growth in all eukaryotes and because of its activation not only in hamartoma syndromes, but in many types of cancer, there is a growing interest in rapamycin and its analogs. In 1999 rapamycin was approved by the Food and Drug Administration (FDA) for the prevention of renal allograft rejection. Currently, clinical studies demonstrate that rapamycin and its analogs, such as CCI-779 (cell cycle inhibitor 779, also known as temsirolimus; Wyeth), RAD001 (also known as everolimus; Novartis), AP23573 (Ariad Pharmaceuticals) have shown anti-cancer activity in variety of malignancies including non-small cell lung carcinoma, advanced neuroendocrine carcinomas, breast, cervical, renal and uterine carcinomas, sarcoma, mesothelioma, metastatic melanoma, relapsed mantle cell lymphoma, glioblastomas and hepatocellular and cholangiocellular cancers.8,127–141 Furthermore, a number of rapamycin analogs are in preclinical development and they are on the way towards the clinic; numerous patents of compounds as PI3K, Akt and mTORC1 inhibitors also have been filed.
Originally, Dr. Krymskaya’s group reported that the mTOR kinase inhibitor rapamycin selectively blocks the constitutive activation of S6K1 and proliferation of cells with mutated TSC2 from LAM patients.12,142 These initial reports have now been confirmed by other laboratories using Eker rat and the TSC1 and TSC2 mouse tumors models.143–145 These pre-clinical studies led to a Phase 2 rapamycin clinical trial for LAM patients with TSC at the Children’s Hospital Medical Center in Cincinnati; the results of these clinical study had been recently published.6 The nonrandomized, open-label Cincinnati Angiomyolipoma Sirolimus Trial for patients with TSC and sporadic LAM demonstrated that treatment with rapamycin leads to a 50% reduction in angiomyolipomas (AML) tumor size and approximately 10% improvement of the lung function in patients with LAM over the first year of treatment.6 Additionally, a case report provided by the Crestani group from Bichat-Claude Bernard Hospital, France, demonstrated that rapamycin treatment of TSC patient with giant angiolipomas induced a dramatic reduction in bilateral renal tumors.9 Although the improvements in AML volume and lung function waned after therapy withdrawal, the initial response suggests that targeting the mTORC1 pathway has promise in the treatment of TSC and LAM.6 Regression of TSC-associated subependymal giant-cell astrocytomas in response to off-label therapy with rapamycin was also reported by Franz group from Cincinnati Children’s Hospital Medical Center.7 On the basis of these studies, a 3-year randomized controlled MILES (Multicenter International LAM Efficacy of Sirolimus) Trial opened in 2006 in Cincinnati. Furthermore, a number of mTOR inhibitor trials for TSC and LAM patients are open for enrollment at the Dana-Farber Cancer Institute, Boston (Phase II clinical trial of sirolimus), Children’s Hospital Medical Center, Cincinnati (Phase I and phase II clinical trials of RED001, Novartis), and the United Kingdom (Phase II clinical trial of sirolimus, Cardiff University, University of Nottingham, St. Georges Hospital Medical School, Royal Sussex County Hospital, Wales Gene Park, The Tuberous Sclerosis Association, and Wyeth).146
Currently, the phase II clinical trial of rapamycin in patients with Cowden syndrome is open for enrollment at the Warren Grant Magnuson Clinical Center, Maryland (NCI, ClinicalTrials.gov Identifier NCT00722449).
Small molecule PI3K inhibitors
Specific, drug-targeted inhibition of PI3K may have beneficial effects for the combination treatment of hamartoma syndromes because it may alleviate the influence of prolonged rapamycin-induced inhibition of mTORC1 on PI3K/Akt activation. Extensive studies of the PI3K family demonstrated that four different, but closely related, PI3K isoforms have distinct biological functions suggesting that specific drug targeting of PI3K may have differential physiological effects.147 PI3Kα plays an important role in tumorigenesis, insulin signaling and glucose metabolism,35,36 PI3Kβ plays a role in thrombosis-related diseases,148 while PI3Kγ and PI3Kδ play key roles in inflammation and the immune system.37,39
While the first generation of pan-PI3K inhibitors, LY294002 (Lilly Research Laboratories),149 wortmannin, a fungal product isolated from Penicillium wortmanni (KY12420, Kyowa Hakko Kogyo Co., Ltd., Japan)150 and wortmannin analogue, PX-866, (Arizona Cancer Center, University of Arizona),151 had been reported to show anticancer effects in vivo, these molecules were not used in clinical trials because of their low specificity and high toxic effects.151,152 During the last 5 years, great progress has been made in the pharmaceutical development of PI3K inhibitors. Novel pan-PI3K inhibitors, which have been developed for cancer therapy, are shown to have anti-tumor activity in cellular and xenograft models (e.g., NVP-BEZ235 (Novartis) possessed strong antiproliferative activity in human prostate tumor PC3M and glioblastoma U87MG cells,153 glioblastoma tumor model,153 and breast cancer xeno-grafts;154 PI-103 (Piramed Pharma) showed significant anti-tumor activity in orthotopic breast and ovarian cancer xenograft models155 as well as in glioma cell models and xenografted EGFR-driven glioma tumors;156 SF1126 (Semafore Pharmaceuticals, Inc.,) inhibited U87MG and PC3 tumor growth;157 ZSTK474 (Zenyaku Kogyo, Japan) inhibited growth of mouse B16F10 melanoma tumors and A549, PC-3 and WiDr human xenografts, which originated from a non-small-cell lung cancer, a prostate cancer and a colon cancer, respectively,158 and XL765 and XL147 (Exelixis) have shown anti-tumor activity in several preclinical xenograft models (information from http://www.exelixis.com). Importantly, novel pan-PI3K inhibitors demonstrate modest toxicity in pre-clinical studies, even though the molecules may cross-react with other members of the PI3K family and PI3K-related kinases.29,147 Because of promising pre-clinical data, some of the second generation pan-PI3K inhibitors have recently entered clinical studies. BEZ235 (Novartis) is now in phase I clinical trial in solid tumors and advanced breast cancer; BGT266 (Novartis) is in ongoing phase I/II trial as monotherapy for breast cancer, solid tumors and Cowden Syndrome. XL765 and XL147 (Exelixis) entered phase I trials in adults with solid tumors and malignant gliomas.
Although broad-range PI3K inhibitors show promising results as anti-cancer agents, selectively targeting individual PI3K isoforms may be also beneficial. Pre-clinical studies suggest that simultaneous inhibition of all PI3K isoforms, which have different functions,35–37,39,148 may lead to a number of pathologies elicited from the modification of normal isoform activity (reviewed in ref. 159), and inactivation of individual PI3K isoforms reduce potential undesirable effects of pan-PI3K inhibitor compounds.
The possibility of developing isoform-selective PI3K inhibitors have been demonstrated by determination of the crystal structure of PI3Kγ. It was shown that minor differences exist in the ATP binding pockets; this allows the production of selective kinase inhibitors.160 Recently, a number of patent specifications have been published which represent promising lead compounds for the future development of isoform-specific PI3K inhibitors: several imidazopyridine derivatives which exhibit excellent PI3K inhibitory activities, especially against p110α, were reported by Yamanouchi;161 quinolone and pyridopyrimidine compounds that are approximately 100-fold more potent against p110α and p110β isoforms compared to PI3Kγ was reported by Thrombogenix;162 number of p110δ quinazoline inhibitors, which inhibit p110δ with 50-fold selectivity over the other class I PI3K isoforms, were reported by ICOS Corporation.163 TG100-115 (TargeGen), a PI3Kγ/δ isoforms-specific inhibitor, was shown to counteract edema and inflammation via blocking VEGF signaling events central to late tissue damage after myocardial infarction. Importantly, preclinical studies show that TG100-115 selectively blocked PI3K/Akt/mTORC1/S6K1 signaling, but not c-Fos and ERK1/2, which were blocked by pan-PI3K inhibitors. Additionally, TG100-115 had no influence on growth, while pan-isoform inhibitors were profoundly anti-proliferative.164 Currently, TG100-115 is in clinical phase I/II trials for the reduction of acute myocardial infarction.164 Given the key role of PI3Kγ in cell signaling, selective inhibitors of PI3Kγ might be also useful in preventing inflammatory cell recruitment in a range of inflammatory diseases, including asthma, rheumatoid arthritis, multiple sclerosis and inflammatory bowel disease. While much of the information concerning more recently developed inhibitors is confined to the patent literature, a side-by-side comparison and activity mapping of PI3K inhibitors is presented in ref.28 The availability of selective PI3K inhibitors provides opportunities for the combination therapy of hamartoma syndromes, supported by recent data. Thus, PI3K inhibitors have been shown to reverse resistance to trastuzumab (herceptin, a monoclonal antibody directed against ERBB2), which was caused by the loss of PTEN.165 Furthermore, novel design-based approach was used to develop dual inhibitor PP121 that targets both PI3K and tyrosine kinase activity.166,167
PI3K/mTORC1 activation: Therapeutic prospects
While the critical role of the mTORC1 signaling pathway proves it is a promising therapeutic target to treat hamartoma syndromes associated with PTEN, LKB1 and TSC1/TSC2 germline mutations, recent studies suggest that caution should be taken in the use of mTOR inhibitors to treat tumors that result from the activation of the mTORC1 signaling pathway due to signaling feedbacks and crosstalks (reviewed in ref. 168). The continuous activation of mTORC1 in cultured cells suppresses insulin-dependent activation of PI3K by downregulating IRS1 (insulin receptor substrate-1) gene expression or by inhibitory phosphorylation of IRS-1.80 Similarly, in vivo, tumors that arise in mice due to loss of TSC2 exhibit constitutive activation of the mTORC1 pathway but not activation of the PI3K pathway.169 This feedback appears to explain why tumors that lose TSC2 are predominantly benign. However, the constitutive activation of the mTORC1 signaling pathway due to TSC2 loss in the context of PI3K activation induces much more aggressive tumors leading to the death of the mice,169 and prolonged treatment with rapamycin may lead to the development of drug-resistance.170,171 Additionally, the substantial pre-clinical data in testing the effects of rapamycin for the treatment of different types of cancer also indicates that rapamycin and its analogs, while having anti-tumor effects, have not shown universal anti-tumor activity in clinical trials.81
Since constitutive activation of mTORC1 provides negative feedback to PI3K/Akt, mTORC1 inhibition may lead to reactivation of PI3K-Akt and higher tumor severity. While pre-clinical study demonstrates that prolonged rapamycin treatment suppresses Akt/PKB signaling due to the destabilization of mTORC2 in some types of cultured cancer cells,172 the results from recent clinical trials provide evidence about PI3K-Akt activation in patient tumors treated with rapamycin or the rapamycin analog RAD001,50,51 Thus, Dr. Rosen’s group demonstrated that mTOR inhibition with RAD001 treatment induced IRS-1 expression and Akt activation in patient tumors.50 Another study shows that inhibition of mTORC1 with RAD001 leads to activation of the MAPK pathway through a PI3K-dependent feedback loop in biopsy-assessed tumor samples.51
Collectively, this evidence demonstrates that the efficiency of the rapamycin family of compounds in preclinical and clinical studies is less then satisfying. Given that over-activation of mTORC1 results in negative feedbacks, it is evident that the use of rapamycin as a single therapeutic agent is limited. Therefore, combinational targeting of PI3K, mTORC1 and MAPK pathways may represent a major direction in treatment of hamartoma syndromes. Given the critical importance of PI3K, mTORC1 and MAPK signaling in normal cellular functions, combinational therapy targeting this pathways may have some limitations due to high toxicity of targeting critical functions in non-cancerous cells. Another possibility could be development of so-called “dirty drugs” that can simultaneously target multiple molecules. For example Novartis and Exelixis developed compounds NVP-BEZ235 and XL765, respectively, for combined targeting of mTOR kinase and PI3K activities.173 However, preclinical studies and the clinical efficacy and tolerability of modulators of PI3K and mTORC1 signaling are eagerly awaited.
Conclusion
The activation of mTORC1 signaling due to loss of function of tumor suppressors PTEN, TSC1/TSC2 and LKB1 in PTEN-related hamartoma syndromes, TSC and PJS make mTORC1 an optimal target for therapy. Clinical trials testing the anti-tumor activity of rapamycin analogs, which specifically inhibit mTORC1 signaling, in patients with TSC/LAM and PTEN-deficient glioblastoma demonstrate promising treatment benefits. These studies also demonstrated the limitations of single drug targeting of mTORC1 with rapamycin, indicating that combinational therapy is required for significant clinical impact. Current developments in specific targeting of PI3K and dual targeting of PI3K and mTORC1 indicate greater promise in finding and developing effective therapy for hamartoma syndromes
Acknowledgments
Supported by grants from NIH/NHLBI 2ROIHL071106 (V.P.K.), 1ROIHL09829 (V.P.K.) and from ATS LAM-07-001(EAG) and Parker B. Francis Fellowship (E.A.G.).
Abbreviations
- 4E-BP1
4E binding protein 1
- AML
angiomyolipoma
- AMPK
AMP-activated protein kinase
- BRRC
bannayan-riley-ruvalcaba syndrome
- CCI-779
cell cycle inhibitor 779
- CS
cowden syndrome
- ERK
extracellular signal-regulated kinases
- FBXW7
F-box and WD repeat domain containing 7
- FDA
food and drug administration
- FKBP
FK506-binding protein
- GAP
GTPase activating protein
- GPCR
G protein coupled receptor
- GSK3
glycogen synthase kinase 3
- IRS1
insulin receptor substrate-1
- LAM
lymphangioleiomyomatosis
- LOH
loss of heterozygosity
- mTOR
mammalian target of rapamycin
- mTORC1
mammalian target of rapamycin complex 1
- mTORC2
mammalian target of rapamycin complex 2
- mSIN1
mammalian stress-activated protein kinase-interaction protein 1
- PDGF
platelet-derived growth factor
- PDK2
phosphoinositide-dependent kinase 2
- PI3K
phosphatidylinositol 3-kinase
- PIP2
phosphatidylinositol-4,5-biphosphate
- PIP3
phosphatidylinositol-3,4,5-trisphosphate
- PJS
peutz-jeghens syndrome
- PLS
proteus-like syndrome
- PRAS40
proline-rich Akt substrate of 40 kDa
- PS
proteus syndrome
- PTEN
phosphatases and tensin homolog
- Raptor
regulatory-associated protein of mTOR
- Rheb
Ras-related small G protein Ras homologue enriched in brain
- RSK
ribosomal protein S6 kinase
- S6K1
p70 ribosomal protein S6 kinase 1
- STK11
serine-threonine kinase 11
- TCTP
translationally controlled tumor protein
- TSC
tuberous sclerosis complex syndrome
- TSC1
tuberous sclerosis complex 1
- TSC2
tuberous sclerosis complex 2
References
- 1.Johnson SR. Lymphangioleiomyomatosis. Eur Respir J. 2006;27:1056–65. doi: 10.1183/09031936.06.00113303. [DOI] [PubMed] [Google Scholar]
- 2.Taveira-DaSilva AM, Steagall WK, Moss J. Lymphangioleiomyomatosis. Cancer Control. 2006;13:276–85. doi: 10.1177/107327480601300405. [DOI] [PubMed] [Google Scholar]
- 3.Crino PB, Nathanson KL, Henske EP. The Tuberous Sclerosis Complex. N Engl J Med. 2006;355:1345–56. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- 4.Krymskaya VP. Smooth muscle-like cells in lymphangioleiomyomatosis. Proc Natl Acad Sci USA. 2007;5:119–26. doi: 10.1513/pats.200705-061VS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goncharova EA, Krymskaya VP. Pulmonary Lymphangioleiomyomatosis (LAM): Progress and Current Challenges. J Cell Biochem. 2008;103:369–82. doi: 10.1002/jcb.21419. [DOI] [PubMed] [Google Scholar]
- 6.Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard JM, et al. Sirolimus for Angiomyolipoma in Tuberous Sclerosis Complex or Lymphangioleiomyomatosis. N Engl J Med. 2008;358:140–51. doi: 10.1056/NEJMoa063564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Franz DN, Leonard J, Tudor C, Chuck G, Care M, Sethuraman G, et al. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol. 2006;59:490–8. doi: 10.1002/ana.20784. [DOI] [PubMed] [Google Scholar]
- 8.Cloughesy TF, Yoshimoto K, Nghiemphu P, Brown K, Dang J, Zhu S, et al. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008;5:8. doi: 10.1371/journal.pmed.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herry I, Neukirch C, Debray MP, Mignon F, Crestani B. Dramatic effect of sirolimus on renal angiomyolipomas in a patient with tuberous sclerosis complex. Eur J Intern Med. 2007;18:76–7. doi: 10.1016/j.ejim.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 10.Consortium. Identification and characterisation of the tuberous sclerosis gene on chromosome 16. Cell. 1993;75:1305–15. doi: 10.1016/0092-8674(93)90618-z. [DOI] [PubMed] [Google Scholar]
- 11.van Slegtenhorst M, Hoogt R, Hermans C, Nellist M, Janssen B, Verhoef S, et al. Identification of the Tuberous Sclerosis Gene TSC1 on Chromosome 9q34. Science. 1997;277:805–8. doi: 10.1126/science.277.5327.805. [DOI] [PubMed] [Google Scholar]
- 12.Goncharova EA, Goncharov DA, Eszterhas A, Hunter DS, Glassberg MK, Yeung RS, et al. Tuberin regulates p70 S6 kinase activation and ribosomal protein S6 phosphorylation: a role for the TSC2 tumor suppressor gene in pulmonary lymphangioleiomyomatosis. J Biol Chem. 2002;277:30958–67. doi: 10.1074/jbc.M202678200. [DOI] [PubMed] [Google Scholar]
- 13.Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, et al. A serine/ threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391:184–7. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- 14.Jenne DE, Reimann H, Nezu J, Friedel W, Loff S, Jeschke R, et al. Peutz-Jeghers syndrome is caused by mutations in a novel serine threoninekinase. Nat Genet. 1998;18:38–43. doi: 10.1038/ng0198-38. [DOI] [PubMed] [Google Scholar]
- 15.Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor supressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 16.Wu X, Senechal K, Neshat MS, Whang YE, Sawyers CR. The PTEN/MMAC1 tumor suppressor phosphatase functions as a negative regulator of phosphatidylinositol 3-kinase/Akt pathway. Proc Natl Acad Sci USA. 1998;95:15587–91. doi: 10.1073/pnas.95.26.15587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liaw D, Marsh DJ, Li J, Dahia PLM, Wang SI, Zheng Z, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1997;16:64–7. doi: 10.1038/ng0597-64. [DOI] [PubMed] [Google Scholar]
- 18.Zhou X-P, Hampel H, Thiele H, Gorlin RJ, Hennekam RCM, Parisi M, et al. Association of germline mutation in the PTEN tumour suppressor gene and Proteus and Proteus-like syndromes. Lancet. 2001;358:210–1. doi: 10.1016/s0140-6736(01)05412-5. [DOI] [PubMed] [Google Scholar]
- 19.Zhou X-P, Marsh DJ, Hampel H, Mulliken JB, Gimm O, Eng C. Germline and germline mosaic PTEN mutations associated with a Proteus-like syndrome of hemihypertrophy, lower limb asymmetry, arteriovenous malformations and lipomatosis. Hum Mol Genet. 2000;9:765–8. doi: 10.1093/hmg/9.5.765. [DOI] [PubMed] [Google Scholar]
- 20.Maehama v, Dixon JE. PTEN: a tumor suppressor that functions as a phospholipid phosphatase. Trends Cell Biol. 1999;9:125–8. doi: 10.1016/s0962-8924(99)01519-6. [DOI] [PubMed] [Google Scholar]
- 21.Inoki K, Corradetti MN, Guan K-L. Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet. 2005;37:19–24. doi: 10.1038/ng1494. [DOI] [PubMed] [Google Scholar]
- 22.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The Biology of Cancer: Metabolic Reprogramming Fuels Cell Growth and Proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 23.Avruch J, Hara K, Lin Y, Liu M, Long X, Ortiz-Vega S, et al. Insulin and amino-acid regulation of mTOR signaling and kinase activity through the Rheb GTPase. Oncogene. 2006;25:6361–72. doi: 10.1038/sj.onc.1209882. [DOI] [PubMed] [Google Scholar]
- 24.Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424–30. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- 25.Lee CH, Inoki K, Guan K. mTOR Pathway as a Target in Tissue Hypertrophy. Annu Rev Pharmacol Toxicol. 2007;47:443–67. doi: 10.1146/annurev.pharmtox.47.120505.105359. [DOI] [PubMed] [Google Scholar]
- 26.Krymskaya VP. Targeting phosphatidylinositol 3-kinase pathway in airway smooth muscle: rationale and promise. BioDrugs. 2007;21:85–95. doi: 10.2165/00063030-200721020-00003. [DOI] [PubMed] [Google Scholar]
- 27.Alessi DR, Sakamoto K, Bayascas JR. LKB1-Dependent Signaling Pathways. Annu Rev Biochem. 2006;75:137–63. doi: 10.1146/annurev.biochem.75.103004.142702. [DOI] [PubMed] [Google Scholar]
- 28.Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, et al. A Pharmacological Map of the PI3-K Family Defines a Role for p110[alpha] in Insulin Signaling. Cell. 2006;125:733–47. doi: 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wymann MP, Schneiter R. Lipid signalling in disease. Nat Rev Mol Cell Biol. 2008;9:162–76. doi: 10.1038/nrm2335. [DOI] [PubMed] [Google Scholar]
- 30.Whitman M, Downes CP, Keeler M, Keller T, Cantley LC. Type 1 phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature. 1988;332:644–6. doi: 10.1038/332644a0. [DOI] [PubMed] [Google Scholar]
- 31.Whitman M, Kaplan D, Roberts T, Cantley L. Evidence for two distinct phosphatidylinositol kinases in fibroblasts. Implications for cellular regulation. Biochem J. 1987;247:165–74. doi: 10.1042/bj2470165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Whitman M, Kaplan DR, Schaffhausen B, Cantley LC, Roberts TM. Association of phosphatidylinositol kinase activity with polyoma middle-T competent for transformation. Nature. 1985;315:239–42. doi: 10.1038/315239a0. [DOI] [PubMed] [Google Scholar]
- 33.Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, et al. Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem. 2001;70:535–602. doi: 10.1146/annurev.biochem.70.1.535. [DOI] [PubMed] [Google Scholar]
- 34.Vanhaesebroeck B, Alessi DR. The PI3K-PDK1 connection: more than just a road to PKB. Biochem J. 2000;346:561–76. [PMC free article] [PubMed] [Google Scholar]
- 35.Bader AG, Kang SA, Zhao L, Vogt PK. Oncogenic PI3K deregulates transcription and translation. Nat Rev Cancer. 2005;5:921–9. doi: 10.1038/nrc1753. [DOI] [PubMed] [Google Scholar]
- 36.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–19. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 37.Chantry D, Vojtek A, Kashishian A, Holtzman D, Wood C, Gray PW, et al. p110δ, a novel phosphatidylinositol 3-kinase catalytic subunit that associates with p85 and is expressed predominantly in leukocytes. J Biol Chem. 1997;272:19236–41. doi: 10.1074/jbc.272.31.19236. [DOI] [PubMed] [Google Scholar]
- 38.Stoyanov B, Volonia S, Hanck T, Rubio I, Loubtchenkov M, Malek D, et al. Cloning and characterization of a G protein-activated human phosphoinositide 3-kinase. Science. 1995;269:690–3. doi: 10.1126/science.7624799. [DOI] [PubMed] [Google Scholar]
- 39.Alcazar I, Marques M, Kumar A, Hirsch E, Wymann M, Carrera AC, et al. Phosphoinositide 3 kinase γ participates in T cell receptor induced T cell activation. J Exp Med. 2007;204:2977–87. doi: 10.1084/jem.20070366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Domin J, Gaidarov I, Smith M, Keen JH, Waterfield MD. The class II phosphoinositide 3-kinase PI3K-C2α is concentrated in the trans-Golgi network and present in clatrin-coated vesicles. J Biol Chem. 2000;275:11943–50. doi: 10.1074/jbc.275.16.11943. [DOI] [PubMed] [Google Scholar]
- 41.Volinia S, Dhand R, Vanhaesebroeck B, MacDougall L, Stein R, Zvelebil MJ, et al. A human phosphatidylinositol 3-kinase complex related to the yeast Vps34p-Vps15p protein sorting system. EMBO J. 1995;14:3339–48. doi: 10.1002/j.1460-2075.1995.tb07340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sansal I, Sellers WR. The Biology and Clinical Relevance of the PTEN Tumor Suppressor Pathway. J Clin Oncol. 2004;22:2954–63. doi: 10.1200/JCO.2004.02.141. [DOI] [PubMed] [Google Scholar]
- 43.Philp AJ, Campbell IG, Leet C, Vincan E, Rockman SP, Whitehead RH, et al. The Phosphatidylinositol 3′-kinase p85{alpha} Gene Is an Oncogene in Human Ovarian and Colon Tumors. Cancer Res. 2001;61:7426–9. [PubMed] [Google Scholar]
- 44.Jücker M, Südel K, Horn S, Sickel M, Wegner W, Fiedler W, et al. Expression of a mutated form of the p85alpha regulatory subunit of phosphatidylinositol 3-kinase in a Hodgkin’s lymphoma-derived cell line (CO) Leukemia. 2002;16:894–901. doi: 10.1038/sj.leu.2402484. [DOI] [PubMed] [Google Scholar]
- 45.Baybis M, Yu J, Lee A, Golden JA, Weiner H, McKhann GI, et al. mTOR cascade activation distinguishes tubers from focal cortical dysplasia. Ann Neurol. 2004;56:478–87. doi: 10.1002/ana.20211. [DOI] [PubMed] [Google Scholar]
- 46.Ma L, Teruya-Feldstein J, Bonner P, Bernardi R, Franz DN, Witte D, et al. Identification of S664 TSC2 Phosphorylation as a Marker for Extracellular Signal-Regulated Kinase Mediated mTOR Activation in Tuberous Sclerosis and Human Cancer. Cancer Res. 2007;67:7106–12. doi: 10.1158/0008-5472.CAN-06-4798. [DOI] [PubMed] [Google Scholar]
- 47.Vézina C, Kudelski A, Sehgal SN. Rapamycin (AY-22,989), a new antifungal antibiotic I. Taxonomy of the producing streptomycete and isolation of the active principle. J Antibiot (Tokyo) 1975;28:721–6. doi: 10.7164/antibiotics.28.721. [DOI] [PubMed] [Google Scholar]
- 48.Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. RAFT1: A mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell. 1994;78:35–43. doi: 10.1016/0092-8674(94)90570-3. [DOI] [PubMed] [Google Scholar]
- 49.Brown EJ, Beal PA, Keith CT, Chen J, Bum Shin T, Schreiber SL. Control of p70 S6 kinase by kinase activity of FRAP in vivo. Nature. 1995;377:441–6. doi: 10.1038/377441a0. [DOI] [PubMed] [Google Scholar]
- 50.O’Reilly KE, Rojo F, She Q, Solit D, Mills GB, Smith D, et al. mTOR Inhibition Induces Upstream Receptor Tyrosine Kinase Signaling and Activates Akt. Cancer Res. 2006;66:1500–8. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065–74. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, et al. mTOR Interacts with Raptor to Form a Nutrient-Sensitive Complex that Signals to the Cell Growth Machinery. Cell. 2002;110:163–75. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 53.Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, et al. Raptor, a Binding Partner of Target of Rapamycin (TOR), Mediates TOR Action. Cell. 2002;110:177–89. doi: 10.1016/s0092-8674(02)00833-4. [DOI] [PubMed] [Google Scholar]
- 54.Kim DH, Sarbassov DD, Ali SM, Latek RR, Guntur KVP, Erdjument-Bromage H, et al. G[beta]L, a Positive Regulator of the Rapamycin-Sensitive Pathway Required for the Nutrient-Sensitive Interaction between Raptor and mTOR. Mol Cell. 2003;11:895–904. doi: 10.1016/s1097-2765(03)00114-x. [DOI] [PubMed] [Google Scholar]
- 55.Schalm SS, Fingar DC, Sabatini DM, Blenis J. TOS Motif-Mediated Raptor Binding Regulates 4E-BP1 Multisite Phosphorylation and Function. Curr Biol. 2003;13:797–806. doi: 10.1016/s0960-9822(03)00329-4. [DOI] [PubMed] [Google Scholar]
- 56.Carrière A, Cargnello M, Julien L, Gao H, Bonneil É, Thibault P, et al. Oncogenic MAPK Signaling Stimulates mTORC1 Activity by Promoting RSK-Mediated Raptor Phosphorylation. Curr Biol. 2008;18:1269–77. doi: 10.1016/j.cub.2008.07.078. [DOI] [PubMed] [Google Scholar]
- 57.Clevers H. Wnt/β-Catenin Signaling in Development and Disease. Cell. 2006;127:469–80. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 58.Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, et al. PRAS40 Is an Insulin-Regulated Inhibitor of the mTORC1 Protein Kinase. Mol Cell. 2007;25:903–15. doi: 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 59.Haar EV, Lee S, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9:316–23. doi: 10.1038/ncb1547. [DOI] [PubMed] [Google Scholar]
- 60.Wang L, Harris TE, Roth RA, Lawrence JC., Jr PRAS40 Regulates mTORC1 Kinase Activity by Functioning as a Direct Inhibitor of Substrate Binding. J Biol Chem. 2007;282:20036–44. doi: 10.1074/jbc.M702376200. [DOI] [PubMed] [Google Scholar]
- 61.Wang L, Harris TE, Lawrence JC., Jr Regulation of Proline-rich Akt Substrate of 40 kDa (PRAS40) Function by Mammalian Target of Rapamycin Complex 1 (mTORC1)-mediated Phosphorylation. J Biol Chem. 2008;283:15619–27. doi: 10.1074/jbc.M800723200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Oshiro N, Takahashi R, Yoshino K, Tanimura K, Nakashima K, Eguchi S, et al. The proline-rich Akt substrate of 40 kDa (PRAS40) is a physioiogical substrate of Mammalian Target of Rapamycin Complex 1. J Biol Chem. 2007;282:20329–39. doi: 10.1074/jbc.M702636200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guertin DA, Sabatini DM. Defining the Role of mTOR in Cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 64.Kozma SC, Thomas G. Regulation of cell size in growth, development and human disease: PI3K, PKB and S6K. BioEssays. 2002;24:65–71. doi: 10.1002/bies.10031. [DOI] [PubMed] [Google Scholar]
- 65.Wullschleger S, Loewith R, Hall MN. TOR Signaling in Growth and Metabolism. Cell. 2006;124:471–84. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 66.Yang Q, Guan K. Expanding mTOR signaling. Cell Res. 2007;17:666–81. doi: 10.1038/cr.2007.64. [DOI] [PubMed] [Google Scholar]
- 67.Zhang Y, Gao X, Saucedo LJ, Ru B, Edgar BA, Pan D. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat Cell Biol. 2003;5:578–81. doi: 10.1038/ncb999. [DOI] [PubMed] [Google Scholar]
- 68.Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, Stocker H, et al. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell. 2003;11:1457–66. doi: 10.1016/s1097-2765(03)00220-x. [DOI] [PubMed] [Google Scholar]
- 69.Castro AF, Rebhun JF, Clark GJ, Quilliam LA. Rheb Binds Tuberous Sclerosis Complex 2 (TSC2) and Promotes S6 Kinase Activation in a Rapamycin- and Farnesylation-dependent Manner. J Biol Chem. 2003;278:32493–6. doi: 10.1074/jbc.C300226200. [DOI] [PubMed] [Google Scholar]
- 70.Inoki K, Li Y, Xu T, Guan K. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17:1829–34. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous Sclerosis Complex Gene Products, Tuberin and Hamartin, Control mTOR Signaling by Acting as a GTPase-Activating Protein Complex toward Rheb. Curr Biol. 2003;13:1259–68. doi: 10.1016/s0960-9822(03)00506-2. [DOI] [PubMed] [Google Scholar]
- 72.Bai X, Ma D, Liu A, Shen X, Wang QJ, Liu Y, et al. Rheb Activates mTOR by Antagonizing Its Endogenous Inhibitor, FKBP38. Science. 2007;318:977–80. doi: 10.1126/science.1147379. [DOI] [PubMed] [Google Scholar]
- 73.Proud CG. mTOR, Unleashed. Science. 2007;318:926–7. doi: 10.1126/science.1150653. [DOI] [PubMed] [Google Scholar]
- 74.Mao J, Kim I, Wu D, Climent J, Kang HC, DelRosario R, et al. FBXW7 Targets mTOR for Degradation and Cooperates with PTEN in Tumor Suppression. Science. 2008;321:1499–502. doi: 10.1126/science.1162981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, et al. Rictor, a Novel Binding Partner of mTOR, Defines a Rapamycin-Insensitive and Raptor-Independent Pathway that Regulates the Cytoskeleton. Curr Biol. 2004;14:1296–302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 76.Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–8. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- 77.Frias MA, Thoreen CC, Jaffe JD, Schroder W, Sculley T, Carr SA, et al. mSin1 Is Necessary for Akt/PKB Phosphorylation, and Its Isoforms Define Three Distinct mTORC2s. Curr Biol. 2006;16:1865–70. doi: 10.1016/j.cub.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 78.Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, et al. Ablation in Mice of the mTORC Components raptor, rictor, or mLST8 Reveals that mTORC2 Is Required for Signaling to Akt-FOXO and PKC[alpha], but Not S6K1. Dev Cell. 2006;11:859–71. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 79.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and Regulation of Akt/ PKB by the Rictor-mTOR Complex. Science. 2005;307:1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 80.Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166:213–23. doi: 10.1083/jcb.200403069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–34. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- 82.Dong J, Pan D. Tsc2 is not a critical target of Akt during normal Drosophila development. Genes Dev. 2004;18:2479–84. doi: 10.1101/gad.1240504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hsu Y-C, Chern JJ, Cai Y, Liu M, Choi K-W. Drosophila TCTP is essential for growth and proliferation through regulation of dRheb GTPase. Nature. 2007;445:785–8. doi: 10.1038/nature05528. [DOI] [PubMed] [Google Scholar]
- 84.Wang X, Fonseca BD, Tang H, Liu R, Elia A, Clemens MJ, et al. Re-evaluating the roles of proposed modulators of mammalian target of rapamycin complex 1 (mTORC1) signaling. J Biol Chem. 2008 doi: 10.1074/jbc.M803348200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and Functional Inactivation of TSC2 by Erk: Implications for Tuberous Sclerosis and Cancer Pathogenesis. Cell. 2005;121:179–93. doi: 10.1016/j.cell.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 86.Hardie DG. New roles for the LKB1 → AMPK pathway. Curr Opin Cell Biol. 2005;17:167–73. doi: 10.1016/j.ceb.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 87.Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern undrestanding of metabolism. Cell Metab. 2005;1:15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 88.Hardie DG. AMP-Activated Protein Kinase as a Drug Target. Annu Rev Pharmacol Toxicol. 2007;47:185–210. doi: 10.1146/annurev.pharmtox.47.120505.105304. [DOI] [PubMed] [Google Scholar]
- 89.Um SH, D’Alessio D, Thomas G. Nutrient overload, insulin resistance and ribosomal protein S6 kinase 1, S6K1. Cell Metab. 2006;3:393–402. doi: 10.1016/j.cmet.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 90.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol Cell. 2008;30:214–26. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hay N. p53 Strikes mTORC1 by Employing Sestrins. Cell Metab. 2008;8:184–5. doi: 10.1016/j.cmet.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Budanov AV, Karin M. p53 Target Genes Sestrin1 and Sestrin2 Connect Genotoxic Stress and mTOR Signaling. Cell. 2008;134:451–60. doi: 10.1016/j.cell.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, et al. TSC2 Integrates Wnt and Energy Signals via a Coordinated Phosphorylation by AMPK and GSK3 to Regulate Cell Growth. Cell. 2006;126:955–68. doi: 10.1016/j.cell.2006.06.055. [DOI] [PubMed] [Google Scholar]
- 94.Choo AY, Roux PP, Blenis J. Mind the GAP: Wnt Steps onto the mTORC1 Train. Cell. 2006;126:834–6. doi: 10.1016/j.cell.2006.08.025. [DOI] [PubMed] [Google Scholar]
- 95.Cheadle JP, Reeve MP, Sampson JR, Kwiatkowski DJ. Molecular genetic advances in tuberous sclerosis. Hum Genet. 2000;107:97–114. doi: 10.1007/s004390000348. [DOI] [PubMed] [Google Scholar]
- 96.Kwiatkowski DJ, Zhang H, Bandura JL, Heiberger KM, Glogauer M, el-Hashemite N, et al. A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and upregulation of p70S6 kinase activity in Tsc1 null cells. Hum Mol Genet. 2002;11:525–34. doi: 10.1093/hmg/11.5.525. [DOI] [PubMed] [Google Scholar]
- 97.Manning BD, Cantley LC. AKT/PKB Signaling: Navigating Downstream. Cell. 2007;129:1261–74. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ozcan U, Ozcan L, Yilmaz E, Dü K, Sahin M, Manning BD, et al. Loss of the Tuberous Sclerosis Complex Tumor Suppressors Triggers the Unfolded Protein Response to Regulate Insulin Signaling and Apoptosis. Mol Cell. 2008;29:541–51. doi: 10.1016/j.molcel.2007.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jiang H, Kenerson H, Aicher L, Miyaoka R, Eary J, Bissler J, et al. The Tuberous Sclerosis Complex Regulates Trafficking of Glucose Transporters and Glucose Uptake. Am J Pathol. 2008;172:1748–56. doi: 10.2353/ajpath.2008.070958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhang H, Bajraszewski N, Wu E, Wang H, Moseman AP, Dabora SL, et al. PDGFRs are critical for PI3K/Akt activation and negatively regulated by mTOR. J Clin Invest. 2007;117:730–8. doi: 10.1172/JCI28984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lee CH, Inoki K, Karbowniczek M, Petroulakis E, Sonenberg N, Henske EP, et al. Constitutive mTOR activation in TSC mutants sensitizes cells to energy starvation and genomic damage via p53. EMBO J. 2007;26:4812–23. doi: 10.1038/sj.emboj.7601900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Goncharova EA, Goncharov DA, Lim PN, Noonan D, Krymskaya VP. Modulation of cell migration and invasiveness by tumor suppressor TSC2 in Lymphangioleiomyomatosis. Am J Respir Cell Mol Biol. 2006;34:473–80. doi: 10.1165/rcmb.2005-0374OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Goncharova E, Goncharov D, Noonan D, Krymskaya VP. TSC2 modulates actin cytoskeleton and focal adhesion through TSC1-binding domain and the Rac1 GTPase. J Cell Biol. 2004;167:1171–82. doi: 10.1083/jcb.200405130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Finlay GA, Malhowski AJ, Liu Y, Fanburg BL, Kwiatkowski DJ, Toksoz D. Selective Inhibition of Growth of Tuberous Sclerosis Complex 2 Null Cells by Atorvastatin Is Associated with Impaired Rheb and Rho GTPase Function and Reduced mTOR/S6 Kinase Activity. Cancer Res. 2007;67:9878–86. doi: 10.1158/0008-5472.CAN-07-1394. [DOI] [PubMed] [Google Scholar]
- 105.Maehama T, Taylor GS, Dixon JE. PTEN AND MYOTUBULARIN: Novel Phosphoinositide Phosphatases. Annu Rev Biochem. 2001;70:247–79. doi: 10.1146/annurev.biochem.70.1.247. [DOI] [PubMed] [Google Scholar]
- 106.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. PTEN, a Putative Protein Tyrosine Phosphatase Gene Mutated in Human Brain, Breast and Prostate Cancer. Science. 1997;275:1943–7. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 107.Steck PA, Pershouse MA, Jasser SA, Yung WKA, Lin H, Ligon AH, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23. 3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–62. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 108.Ali IU, Schriml LM, Dean M. Mutational Spectra of PTEN/MMAC1 Gene: a Tumor Suppressor With Lipid Phosphatase Activity. J Natl Cancer Inst. 1999;91:1922–32. doi: 10.1093/jnci/91.22.1922. [DOI] [PubMed] [Google Scholar]
- 109.Simpson L, Parsons R. PTEN: Life as a Tumor Suppressor. Exp Cell Res. 2001;264:29–41. doi: 10.1006/excr.2000.5130. [DOI] [PubMed] [Google Scholar]
- 110.Zbuk KM, Eng C. Cancer phenomics: RET and PTEN as illustrative models. Nat Rev Cancer. 2007;7:35–45. doi: 10.1038/nrc2037. [DOI] [PubMed] [Google Scholar]
- 111.Zbuk KM, Eng C. Hamartomatous polyposis syndromes. Nat Clin Pract Gastroenterol Hepatol. 2007;4:492–502. doi: 10.1038/ncpgasthep0902. [DOI] [PubMed] [Google Scholar]
- 112.Gorlin RJ, Cohen MM, Condon LM, Burke BA. Bannayan-Riley-Ruvalcaba syndrome. Am J Med Genet. 1992;44:307–14. doi: 10.1002/ajmg.1320440309. [DOI] [PubMed] [Google Scholar]
- 113.Biesecker L. The challenges of Proteus syndrome: diagnosis and management. Eur J Hum Genet. 2006;14:1151–7. doi: 10.1038/sj.ejhg.5201638. [DOI] [PubMed] [Google Scholar]
- 114.Shaw RJ, Lamia KA, Vasquez DS, Koo SH, Bardeesy N, DePinho RA, et al. The Kinase LKB1 Mediates Glucose Homeostasis in Liver and Therapeutic Effects of Metformin. Science. 2005;310:11642–6. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–90. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 116.Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. 2004;6:91–9. doi: 10.1016/j.ccr.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 117.Corradetti MN, Inoki K, Bardeesy N, DePinho RA, Guan K. Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev. 2004;18:1533–8. doi: 10.1101/gad.1199104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sanchez-Cespedes M. A role for LKB1 gene in human cancer beyond the Peutz-Jeghers syndrome. Oncogene. 2007;26:7825–32. doi: 10.1038/sj.onc.1210594. [DOI] [PubMed] [Google Scholar]
- 119.Knudson AG. Two genetic hits (more or less) to cancer. Nat Rev Cancer. 2001;1:157–62. doi: 10.1038/35101031. [DOI] [PubMed] [Google Scholar]
- 120.Li S, Takeuchi F, Wang J, Fan QW, Komurasaki T, Billings EM, et al. Mesenchymal-epithelial interactions involving epiregulin in tuberous sclerosis complex hamartomas. Proc Natl Acad Sci USA. 2008;105:3539–44. doi: 10.1073/pnas.0712397105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Whang YE, Wu X, Suzuki H, Reiter RE, Tran C, Vessella RL, et al. Inactivation of the tumor suppressor PTEN/MMAC1 in advanced human prostate cancer through loss of expression. Proc Natl Acad Sci USA. 1998;95:5246–50. doi: 10.1073/pnas.95.9.5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Li DM, Sun H. TEP1, Encoded by a Candidate Tumor Suppressor Locus, Is a Novel Protein Tyrosine Phosphatase Regulated by Transforming Growth Factor β. Cancer Res. 1997;57:2124–9. [PubMed] [Google Scholar]
- 123.Waite KA, Eng C. From developmental disorder to heritable cancer: it’s all in the BMP/ TGFβ family. Nat Rev Genet. 2003;4:763–73. doi: 10.1038/nrg1178. [DOI] [PubMed] [Google Scholar]
- 124.Torres J, Pulido R. The Tumor Suppressor PTEN Is Phosphorylated by the Protein Kinase CK2 at Its C Terminus. Implications for PTEN stability to proteasome-mediated degradation. J Biol Chem. 2001;276:993–8. doi: 10.1074/jbc.M009134200. [DOI] [PubMed] [Google Scholar]
- 125.Vazquez F, Ramaswamy S, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail regulates protein stibility and function. Mol Cell Biol. 2000;20:5010–8. doi: 10.1128/mcb.20.14.5010-5018.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hartford CM, Ratain MJ. Rapamycin: Something Old, Something New, Sometimes Borrowed and Now Renewed. Clin Pharmacol Ther. 2007;82:381–8. doi: 10.1038/sj.clpt.6100317. [DOI] [PubMed] [Google Scholar]
- 127.Duran I, Kortmansky J, Singh D, Hirte H, Kocha W, Goss G, et al. A phase II clinical and pharmacodynamic study of temsirolimus in advanced neuroendocrine carcinomas. Br J Cancer. 2006;95:1148–54. doi: 10.1038/sj.bjc.6603419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hidalgo M, Buckner JC, Erlichman C, Pollack MS, Boni JP, Dukart G, et al. A Phase I and Pharmacokinetic Study of Temsirolimus (CCI-779) Administered Intravenously Daily for 5 Days Every 2 Weeks to Patients with Advanced Cancer. Clin Cancer Res. 2006;12:5755–63. doi: 10.1158/1078-0432.CCR-06-0118. [DOI] [PubMed] [Google Scholar]
- 129.Margolin K, Longmate J, Baratta T, Synold T, Christensen S, Weber J, et al. CCI-779 in metastatic melanoma. Cancer. 2005;104:1045–8. doi: 10.1002/cncr.21265. [DOI] [PubMed] [Google Scholar]
- 130.Galanis E, Buckner JC, Maurer MJ, Kreisberg JI, Ballman K, Boni J, et al. Phase II Trial of Temsirolimus (CCI-779) in Recurrent Glioblastoma Multiforme: A North Central Cancer Treatment Group Study. J Clin Oncol. 2005;23:5294–304. doi: 10.1200/JCO.2005.23.622. [DOI] [PubMed] [Google Scholar]
- 131.Witzig TE, Geyer SM, Ghobrial I, Inwards DJ, Fonseca R, Kurtin P, et al. Phase II Trial of Single-Agent Temsirolimus (CCI-779) for Relapsed Mantle Cell Lymphoma. J Clin Oncol. 2005;23:5347–56. doi: 10.1200/JCO.2005.13.466. [DOI] [PubMed] [Google Scholar]
- 132.Chan S, Scheulen ME, Johnston S, Mross K, Cardoso F, Dittrich C, et al. Phase II Study of Temsirolimus (CCI-779), a Novel Inhibitor of mTOR, in Heavily Pretreated Patients With Locally Advanced or Metastatic Breast Cancer. J Clin Oncol. 2005;23:5314–22. doi: 10.1200/JCO.2005.66.130. [DOI] [PubMed] [Google Scholar]
- 133.Boni JP, Leister C, Bender G, Fitzpatrick V, Twine N, Stover J, et al. Population Pharmacokinetics of CCI-779: Correlations to Safety and Pharmacogenomic Responses in Patients with Advanced Renal Cancer. Clin Pharmacol Ther. 2005;77:76–89. doi: 10.1016/j.clpt.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 134.Atkins MB, Hidalgo M, Stadler WM, Logan TF, Dutcher JP, Hudes GR, et al. Randomized Phase II Study of Multiple Dose Levels of CCI-779, a Novel Mammalian Target of Rapamycin Kinase Inhibitor, in Patients With Advanced Refractory Renal Cell Carcinoma. J Clin Oncol. 2004;22:909–18. doi: 10.1200/JCO.2004.08.185. [DOI] [PubMed] [Google Scholar]
- 135.Rizell M, Andersson M, Cahlin C, Hafström L, Olausson M, Lindnér P. Effects of the mTOR inhibitor sirolimus in patients with hepatocellular and cholangiocellular cancer. Int J Clin Oncol. 2008;13:66–70. doi: 10.1007/s10147-007-0733-3. [DOI] [PubMed] [Google Scholar]
- 136.Gore ME. Temsirolimus in the treatment of advanced renal cell carcinoma. Ann Oncol. 2007;18:87–8. doi: 10.1093/annonc/mdm299. [DOI] [PubMed] [Google Scholar]
- 137.Motzer RJ, Hudes GR, Curti BD, McDermott DF, Escudier BJ, Negrier S, et al. Phase I/II Trial of Temsirolimus Combined With Interferon Alfa for Advanced Renal Cell Carcinoma. J Clin Oncol. 2007;25:3958–64. doi: 10.1200/JCO.2006.10.5916. [DOI] [PubMed] [Google Scholar]
- 138.Milton DT, Riely GJ, Azzoli CG, Gomez JE, Heelan RT, Kris MG, et al. Phase 1 trial of everolimus and gefitinib in patients with advanced nonsmall-cell lung cancer. Cancer. 2007;110:599–605. doi: 10.1002/cncr.22816. [DOI] [PubMed] [Google Scholar]
- 139.Garland LL, Rankin C, Gandara DR, Rivkin SE, Scott KM, Nagle RB, et al. Phase II Study of Erlotinib in Patients With Malignant Pleural Mesothelioma: A Southwest Oncology Group Study. J Clin Oncol. 2007;25:2406–13. doi: 10.1200/JCO.2006.09.7634. [DOI] [PubMed] [Google Scholar]
- 140.Okuno S. Mammalian target of rapamycin inhibitors in sarcomas. Curr Opin Oncol. 2006:18. doi: 10.1097/01.cco.0000228742.72165.cf. [DOI] [PubMed] [Google Scholar]
- 141.Vignot S, Faivre S, Aguirre D, Raymond E. mTOR-targeted therapy of cancer with rapamycin derivatives. Ann Oncol. 2005;16:525–37. doi: 10.1093/annonc/mdi113. [DOI] [PubMed] [Google Scholar]
- 142.Goncharova EA, Goncharov DA, Spaits M, Noonan D, Talovskaya E, Eszterhas A, et al. Abnormal Smooth Muscle Cell Growth in Lymphangioleiomyomatosis (LAM): Role for Tumor Suppressor TSC2. Am J Respir Cell Mol Biol. 2006;34:561–72. doi: 10.1165/rcmb.2005-0300OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kenerson HL, Aicher LD, True LD, Yeung RS. Activated mammalian target of rapamycin pathway in the pathogenesis of tuberous sclerosis complex renal tumors. Cancer Res. 2002;62:5645–50. [PubMed] [Google Scholar]
- 144.Lee L, Sudentas P, Donohue B, Asrican K, Worku A, Walker V, et al. Efficacy of a rapamycin analog (CCI-779) and IFNγ in tuberous sclerosis mouse models. Genes Chromosomes Cancer. 2005;42:213–27. doi: 10.1002/gcc.20118. [DOI] [PubMed] [Google Scholar]
- 145.Yeung RS. Multiple roles of the tuberous sclerosis complex genes. Genes Chromosomes Cancer. 2003;38:368–75. doi: 10.1002/gcc.10256. [DOI] [PubMed] [Google Scholar]
- 146.McCormack FX. Lymphangioleiomyomatosis: A Clinical Update. Chest. 2008;133:507–16. doi: 10.1378/chest.07-0898. [DOI] [PubMed] [Google Scholar]
- 147.Marone R, Cmiljanovic V, Giese B, Wymann MP. Targeting phosphoinositide 3-kinase—Moving towards therapy. Biochim Biophys Acta. 2008;1784:159–85. doi: 10.1016/j.bbapap.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 148.Jackson SP, Schoenwaelder SM, Goncalves I, Nesbitt WS, Yap CL, Wright CE, et al. PI 3-kinase p110[beta]: a new target for antithrombotic therapy. Nat Med. 2005;11:507–14. doi: 10.1038/nm1232. [DOI] [PubMed] [Google Scholar]
- 149.Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) J Biol Chem. 1994;269:5241–8. [PubMed] [Google Scholar]
- 150.Okada T, Sakuma L, Fukui Y, Hazeki O, Ui M. Blockage of chemotactic peptide-induced stimulation of neutrophils by wortmannin as a result of selective inhibition of phosphatidylinositol 3-kinase. J Biol Chem. 1994;269:3563–7. [PubMed] [Google Scholar]
- 151.Ihle NT, Williams R, Chow S, Chew W, Berggren MI, Paine-Murrieta G, et al. Molecular pharmacology and antitumor activity of PX-866, a novel inhibitor of phosphoinositide-3-kinase signaling. Mol Cancer Ther. 2004;3:763–72. [PubMed] [Google Scholar]
- 152.Hu L, Zaloudek C, Mills GB, Gray J, Jaffe RB. In vivo and in vitro Ovarian Carcinoma Growth Inhibition by a Phosphatidylinositol 3-Kinase Inhibitor (LY294002) Clin Cancer Res. 2000;6:880–6. [PubMed] [Google Scholar]
- 153.Maira S, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/ mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7:1851–63. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 154.Serra V, Markman B, Scaltriti M, Eichhorn PJA, Valero V, Guzman M, et al. NVP-BEZ235, a Dual PI3K/mTOR Inhibitor, Prevents PI3K Signaling and Inhibits the Growth of Cancer Cells with Activating PI3K Mutations. Cancer Res. 2008;68:8022–30. doi: 10.1158/0008-5472.CAN-08-1385. [DOI] [PubMed] [Google Scholar]
- 155.Raynaud FI, Eccles S, Clarke PA, Hayes A, Nutley B, Alix S, et al. Pharmacologic Characterization of a Potent Inhibitor of Class I Phosphatidylinositide 3-Kinases. Cancer Res. 2007;67:5840–50. doi: 10.1158/0008-5472.CAN-06-4615. [DOI] [PubMed] [Google Scholar]
- 156.Fan QW, Knight ZA, Goldenberg DD, Yu W, Mostov KE, Stokoe D, et al. A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell. 2006;9:341–9. doi: 10.1016/j.ccr.2006.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Garlich JR, De P, Dey N, Su JD, Peng X, Miller A, et al. A Vascular Targeted Pan Phosphoinositide 3-Kinase Inhibitor Prodrug, SF1126, with Antitumor and Antiangiogenic Activity. Cancer Res. 2008;68:206–15. doi: 10.1158/0008-5472.CAN-07-0669. [DOI] [PubMed] [Google Scholar]
- 158.Yaguchi S, Fukui Y, Koshimizu I, Yoshimi H, Matsuno T, Gouda H, et al. Antitumor Activity of ZSTK474, a New Phosphatidylinositol 3-Kinase Inhibitor. J Natl Cancer Inst. 2006;98:545–56. doi: 10.1093/jnci/djj133. [DOI] [PubMed] [Google Scholar]
- 159.Wee S, Lengauer C, Wiederschain D. Class IA phosphoinositide 3-kinase isoforms and human tumorigenesis: implications for cancer drug discovery and development. Curr Opin Oncol. 2008;20:77–82. doi: 10.1097/CCO.0b013e3282f3111e. [DOI] [PubMed] [Google Scholar]
- 160.Walker EH, Pacold ME, Perisic O, Stephens LC, Hawkins PT, Wymann MP, et al. Structural Determinants of Phosphoinositide 3-Kinase Inhibition by Wortmannin, LY294002, Quercetin, Myricetin and Staurosporine. Mol Cell. 2000;6:909–19. doi: 10.1016/s1097-2765(05)00089-4. [DOI] [PubMed] [Google Scholar]
- 161.Hayakaa M, Kaizawa H, Kawaguchi KI, Matsuda K, Ishikawa N, Koizumi T, et al. United States Patent. USA: Yamanouchi Pharmaceutical Co., Ltd; 2002. Preparation of imidazopyridine derivatives as phosphatidylinositol 3-kinase inhibitors and anticancer agents. [Google Scholar]
- 162.Robertson AJ, Jackson S, Kenche V, Yaip C, Parbaharan H, Thompson P. International patent application. 2001. Therapeutic morpholino-substituted compounds. [Google Scholar]
- 163.Sadhu C, Dick K, Treiberg J, Sowell CG, Kesicki EA, Oliver A. International patent application. 2001. Quinazolinone derivatives as inhibitors of human phosphatidylinositol 3-kinase delta. [Google Scholar]
- 164.Doukas J, Wrasidlo W, Noronha G, Dneprovskaia E, Hood J, Soll R. Isoform-selective PI3K inhibitors as novel therapeutics for the treatment of acute myocardial infarction. Biochem Soc Trans. 2007;35:204–6. doi: 10.1042/BST0350204. [DOI] [PubMed] [Google Scholar]
- 165.Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6:117–27. doi: 10.1016/j.ccr.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 166.Apsel B, Blair JA, Gonzalez B, Nazif TM, Feldman ME, Aizenstein B, et al. Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat Chem Biol. 2008;4:691–9. doi: 10.1038/nchembio.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Bilanges B, Torbett N, Vanhaesebroeck B. Killing two kinase families with one stone. Nat Chem Biol. 2008;4:648–9. doi: 10.1038/nchembio1108-648. [DOI] [PubMed] [Google Scholar]
- 168.Carracedo A, Pandolfi PP. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene. 27:5527–41. doi: 10.1038/onc.2008.247. [DOI] [PubMed] [Google Scholar]
- 169.Manning BD, Logsdon MN, Lipovsky AI, Abbott D, Kwiatkowski DJ, Cantley LC. Feedback inhibition of Akt signaling limits the growth of tumors lacking Tsc2. Genes Dev. 2005;19:1773–8. doi: 10.1101/gad.1314605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Dilling MB, Germain GS, Dudkin L, Jayaraman AL, Zhang X, Harwood FC, et al. 4E-binding Proteins, the Suppressors of Eukaryotic Initiation Factor 4E, are downregulated in Cells with Acquired or Intrinsic Resistance to Rapamycin. J Biol Chem. 2002;277:13907–17. doi: 10.1074/jbc.M110782200. [DOI] [PubMed] [Google Scholar]
- 171.Marx SO, Marks AR. Cell cycle progression and proliferation despite 4BP-1 dephosphorylation. Mol Cell Biol. 1999;19:6041–7. doi: 10.1128/mcb.19.9.6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Sarbassov DD, Ali SM, Sengupta S, Sheen J, Hsu PP, Bagley AF, et al. Prolonged Rapamycin Treatment Inhibits mTORC2 Assembly and Akt/PKB. Mol Cell. 2006;22:159–68. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 173.Garcia-Echeverria C, Sellers WR. Drug discovery approaches targeting the PI3K//Akt pathway in cancer. Oncogene. 27:5511–26. doi: 10.1038/onc.2008.246. [DOI] [PubMed] [Google Scholar]